Benchmarking SaladCloud: GPU Performance and Reliability in Action

SaladCloud is powered by tens of thousands of globally distributed nodes—primarily high-performance desktop computers and servers running the SaladCloud agent. Each node is equipped with consumer-grade or data center GPUs, along with diverse CPU and memory configurations. Node distribution varies by region: consumer GPU nodes in North America make up 50–60% of the total, while nearly all data center GPU nodes are currently located in the US. Because of the distributed architecture, nodes can differ significantly in network throughput, startup time, and uptime—all critical factors to consider when designing applications for SaladCloud. In this benchmark, we deployed a workload with 100 replicas over seven days to measure key metrics such as startup times, interruptions and reallocations, uptimes, run-to-request radio and performances. These results provide practical reference points for building high-performance, reliable applications on SaladCloud. Benchmark Methodology The test workload, packaged as a 5.53 GB (compressed) image, is integrated with the GPU-intensive lolMiner. During execution, it collects key metrics every minute, including mining performance (solutions/s), resource utilization (CPU, RAM, GPU, VRAM), and GPU temperature. Aggregated metrics are saved to the cloud every 5 minutes, with approximately 2.5 minutes of data lost during each reallocation. Please refer to the test code for more details. We executed the workload by deploying a container group with 100 replicas, each provisioned with 8 vCPUs, 16 GB of memory, and all available consumer GPU types at high priority. The group ran continuously for 7 days and 2 hours, during which each instance generated a metric file that was stored in the designated bucket and folder in Cloudflare R2. We then downloaded and analyzed these files to produce the key metrics. It is important to note that outcomes can vary depending on factors such as image size, replica count, selected regions and resource types, workload priority, and test duration. Consequently, results may differ under different circumstances. Startup Times Startup time includes instance allocation, image download, and decompression, and is influenced by several factors: Starting at 00:00 UTC on September 22, 2025, we tracked how many instances became operational over time to measure startup times. Key observations include: Interruptions and Reallocations Running instances may go offline for various reasons, in which case new instances are allocated to continue processing: We skipped the first 2 hours of startup, allowing initial allocations to complete. We then monitored how many new instances became operational over time to assess interruptions and reallocations. Key observations are: SaladCloud’s data center nodes can reliably run high-priority workloads without interruptions from their providers. Uptimes Uptime is mainly influenced by interruptions, reallocations, and startup delays. By recording when each instance first came online and its last update, we can calculate the effective uptime. Key observations are: High-priority applications on SaladCloud’s data center nodes generally run uninterrupted for extended periods. Run-to-Request Ratio The instance run-to-request ratio measures the actual compute capacity available compared to what is requested. For example, when requesting 100 instances, the ratio indicates how many are running at any given time. When a node goes offline and a replacement is allocated, additional time is required to download and decompress the image before the new instance becomes operational. Because of variations in startup times and uptimes, a 100% run-to-request ratio cannot be consistently guaranteed on SaladCloud’s consumer GPU nodes. Large image sizes can increase startup times, which in turn lowers the run-to-request ratio. To mitigate this, it is often necessary to provision additional instances (5~10%) beyond the initial plan, particularly for real-time inference workloads. By tracking the number of running instances over time, we can calculate the run-to-request ratio. Key observations include: Processing Performance On SaladCloud, nodes with the same consumer GPU type can exhibit varying performance due to differences in system configuration (CPU, RAM), clock speed, cooling, and power limits. For a single node, its performance may fluctuate over time as a result of temperature changes and cooling efficiency. Based on the test, over 90% of SaladCloud’s consumer nodes deliver stable and consistent performance, and all data center nodes do so. The following instance run illustrates stable performance (black line) and resource usage over the 7-day execution period: To manage performance variances and fluctuations from a small number of consumer GPU nodes, we recommend conducting an initial check and real-time performance monitoring to select suitable nodes and ensure they remain in an optimal state for application execution. Build High-Performance, Reliable Applications on SaladCloud The benchmark test offers insights into the distributed nature of SaladCloud and provides reference data for building high-performance, reliable applications. Applications can be optimized to address specific challenges and fully leverage SaladCloud’s exceptional flexibility and cost effectiveness, enabling GPU-powered workloads to scale up or down at highly competitive prices. For more details, please refer to this guide. For workloads that process millions of jobs in a short time or require access to hundreds of GPUs—including multiple types for testing or research—SaladCloud offers an ideal platform. Deploy a GPU on SaladCloud Today Interested in running your workload on SaladCloud (H100s, A100s, RTX 4090s, and more)? Check out SaladCloud. SaladCloudSaladCloud is the world’s largest distributed cloud computing network with 11,000+ daily GPUs and 450,000 GPUs contributing compute, all at the lowest cost in the market.
Salad Slashes GPU Pricing Again – Now the Serving the Lowest Cost GPUs on the Market

Salad Makes GPU Computing Even More Affordable We’re thrilled to announce another massive round of price cuts across our entire GPU fleet, cementing SaladCloud’s position as the most affordable cloud computing platform in the world. With these new prices, we’re not just competing – we’re redefining what affordable cloud computing looks like. The highlights? You can now rent (on batch priority): And these aren’t just advertised prices. With over 450,000 compute providers and growing daily, SaladCloud delivers unmatched availability at these breakthrough price points. The Numbers Don’t Lie: Salad vs. The Competition Let’s get straight to the point. Here’s how SaladCloud’s new pricing stacks up against the lowest prices we could find from other major cloud providers: Where We Dominate the Competition What Makes These Prices Possible? Our distributed compute network continues to grow exponentially. We now have: This scale advantage means we can offer enterprise-grade GPUs at prices that traditional cloud providers simply cannot match. Four-Tier Pricing: Choose Your Priority Level We’ve enhanced our pricing model with four distinct priority tiers to match your workload requirements: GPU Type High Priority Medium Priority Low Priority Batch Priority H100 NVL (94GB) $1.80 $1.53 $1.26 $0.99 H100 SXM (80GB) $1.80 $1.53 $1.26 $0.99 A100 SXM (80GB) $1.00 $0.83 $0.67 $0.50 A100 PCIe (40GB) $0.95 $0.77 $0.58 $0.40 L40S (48GB) $0.81 $0.65 $0.48 $0.32 RTX 5090 (32GB) $0.45 $0.38 $0.31 $0.25 RTX 5080 (16GB) $0.42 $0.34 $0.25 $0.18 RTX 4090 (24GB) $0.30 $0.25 $0.20 $0.16 Multi-GPU Configurations Available Need serious compute power? We’ve got you covered: When to Choose Each Priority Tier High Priority ($$$): Mission-critical inference serving thousands of users, requiring 24/7 availability. Medium Priority ($$): Production workloads that need reliable uptime but can handle occasional interruptions. Low Priority ($): Development, testing, and non-time-critical batch processing. Batch Priority: Long-running training jobs, research workloads, and cost-sensitive batch processing where maximum savings matter more than guaranteed availability. How to Start Saving Today The Salad Advantage: Why We’re Different Massive Scale: 450,000+ GPUs means we can handle your largest workloads True Availability: Our distributed network provides better uptime than traditional clouds Zero Lock-in: Standard containerized deployments work everywhere Transparent Pricing: No hidden fees, no complex billing structures Enterprise Support: Dedicated support for high-volume users What’s Driving These Price Cuts? Increased Supply: Our network has grown 3x in the past year, driving down acquisition costs Operational Efficiency: Zero infrastructure overhead means maximum savings for customers Market Leadership: We’re not just competing on price – we’re setting the standard Customer Success: Your success drives our growth, creating a virtuous cycle of lower prices Enterprise Pricing Available For workloads requiring 1,000+ GPU hours monthly, custom SLAs, or dedicated support, our enterprise team offers additional volume discounts. Book a consultation: Talk to Sales Deploy a GPU on SaladCloud Today With these new prices, there’s never been a better time to move your compute workloads to SaladCloud. Whether you’re training the next breakthrough AI model, rendering the next blockbuster game, or processing massive datasets, SaladCloud delivers enterprise-grade performance at consumer-friendly prices. Interested in running your workload on SaladCloud (H100s, A100s, RTX 4090s, and more)? Check out SaladCloud. SaladCloudSaladCloud is the world’s largest distributed cloud computing network with 11,000+ daily GPUs and 450,000 GPUs contributing compute, all at the lowest cost in the market.
Generate a Minute of High-Quality AI Video with WAN2.2 on SaladCloud for as Little as $0.87

Over the past two years, the field of AI video generation has exploded. Proprietary systems like OpenAI’s Sora, Runway Gen-2, and Pika have shown the world what’s possible: turning a few words or an image prompt into seconds—or even minutes—of coherent video. But the open-source community hasn’t stood still. A wave of open-source video generation models is quickly closing the gap, giving creators access to powerful tools that support text-to-video, image-to-video, and video-to-video workflows. At SaladCloud, we’re focused on what it takes to run these models in production: how they scale across GPUs, what kind of hardware they require, and what the true cost per minute of generated video looks like. Before we dive into WAN2.2 benchmarks, let’s take a quick look at the open-source landscape. How Open-Source Video Generation Works The majority of open-source video generation models today are based on diffusion or flow-matching techniques—extensions of the same methods that power Stable Diffusion, adapted to generate sequences of frames. Other families of approaches do exist (e.g., GAN-based or autoregressive methods), but diffusion and flow-matching dominate the current open-source landscape due to their scalability and image quality. These models support a variety of creative workflows: Available Open-Source Models When considering which video model to run in production, three things matter most: Here’s a look at six of the most relevant open-source video models today: Model License Parameters Memory (approx) Notes WAN2.1 Apache 2.0 1.3B / 14B 8–60 GB Early release; artifact-heavy, scaling issues. WAN2.2 Apache 2.0 5B / A14B 24-80 GB Popular, controllable, strong ecosystem. Skyreels-V2 Permissive 14B ~42 GB Broad adoption, rich controllability features. LTX Non-permissive 2-13B ~13 GB+ Efficient, comes in size variants for trade-offs. Mochi Apache 2.0 10B ~57 GB Innovative, but adoption has slowed. Nova Apache 2.0 0.3-1.4B ~30 GB Stability AI’s latest; balances quality & efficiency. WAN2.2: The Best Open-Source Video Model Yet When we covered WAN2.1, it showed the possibility of open-source video generation. WAN2.2, however, delivers what’s more practical: cinematic-quality videos generated on hardware you can afford. Highlights of WAN2.2 vs WAN2.1 WAN2.2 is probably the best open-source video generation model available today, transforming AI video from experimental tech into production-ready infrastructure. WAN2.2 comes in two primary variants: WAN2.2 – The 5B Model The WAN2.2-TI2V-5B is the most approachable entry point into WAN2.2: Key Traits of the 5B Model: Benchmark Results – 5B with 1280×704 resolution We ran the 5B model on SaladCloud, using both: All benchmarks used the official WAN2.2 repo and weights provided by WAN, with no CPU offloading or custom optimizations: GPU Type Resolution Avg Gen Time (5s) Time per 1-min Video Cost per Min (High Priority) Cost per Min (Batch Priority) L40S (Secure) 1280×704 13.5 min 162 min $2.19 $0.87 4090 1280×704 31.8 min 381.6 min $2.42 $1.65 5090 1280×704 16.3 min 195.6 min $1.73 $1.14 With the 5B model, you can create a full minute of 1280×704 video for under $1 using L40S Secure GPUs with batch pricing. If you need to scale to hundreds or even thousands of instances SaladCloud consumer GPU’s might be a great option as well. Video generated using WAN2.2 5B model on rtx5090. 1280×704 “Medium shot, stainless modern kitchen, pancakes cooking while steam rises, warm backlight, subtle parallax from foreground utensils, 35mm lens” WAN2.2 – The A14B Model The A14B variant of WAN2.2 pushes video generation to its absolute limits. With 14B active parameters per step and a Mixture-of-Experts architecture, it produces some of the sharpest, most cinematic open-source video today. While trying to run the model benchmark we faced a challenge: the official model requires 80+ GB of VRAM, making it impossible to be hosted on most GPUs. To make benchmarking possible, we used the DFloat11 compressed release. This version applies Huffman coding to the exponent bits of BFloat16 weights, taking advantage of their high compressibility. The algorithm performs on-the-fly weight decompression directly on the GPU, which drastically lowers memory requirements while preserving performance. You can learn more in DFloat11’s research paper on hardware-aware compression. Benchmark Results – A14B We ran the compressed A14B model on SaladCloud, across Secure datacenter GPUs: l40s and A100s. GPU Type Resolution Avg Gen Time (5s) Time per 1-min Video Cost per Min (High Priority) Cost per Min (Batch Priority) L40S 1280×704 55.1 min 11.02 h $8.93 $3.53 A100 1280×704 59.7 min 11.93 h $11.34 $4.77 L40S 480p 14.42 min 2.88 h $2.34 $0.92 A100 480p 14.45 min 2.89 h $2.75 $1.16 WAN2.2 A14B sets the quality bar for open-source video generation. At 1280×704, it is still compute-heavy (10+ hours per 1 min clip), but at 480p it becomes viable for production workflows. For broadcast or cinematic projects, this is the best model up to date. Video generated using WAN2.2 A14B model on L40S. 720×480 “A cinematic scene of a mountain climber reaching the snowy summit” Video generated using WAN2.2 A14B model on A100. 1280×720 “A cinematic scene of a couple walking under cherry blossoms in the wind” The Economics of WAN2.2 on SaladCloud Beyond speed and fidelity, the real breakthrough with WAN2.2 on SaladCloud is cost efficiency. Video generation is notoriously expensive, and for most teams, the economics have been the biggest barrier to actually using these models at scale. By running on Salad’s distributed GPU’s, you get access to both consumer GPUs (4090/5090) and Secure datacenter GPUs (L40S, A100) at prices far below traditional cloud providers which makes SaladCloud the most affordable way to produce a minute of high-quality video with WAN2.2 today. Best-Case Costs per Minute of Finished Video To put that in perspective, generating the same output on a traditional GPU cloud often costs two to five times more. For anyone looking to scale beyond short clips into long-form or professional production, this cost advantage is a game-changer. Interested in running your workload on SaladCloud Secure (H100s, A100s, L40S, and more)? Check out SaladCloud Secure. SaladCloudSaladCloud is the world’s largest distributed cloud computing network with 11,000+ daily GPUs and 450,000 GPUs contributing compute, all at the lowest cost in the market.
Blender Rendering Benchmark across 27 Consumer and Data Center GPUs on SaladCloud, with 90%+ Cost Savings
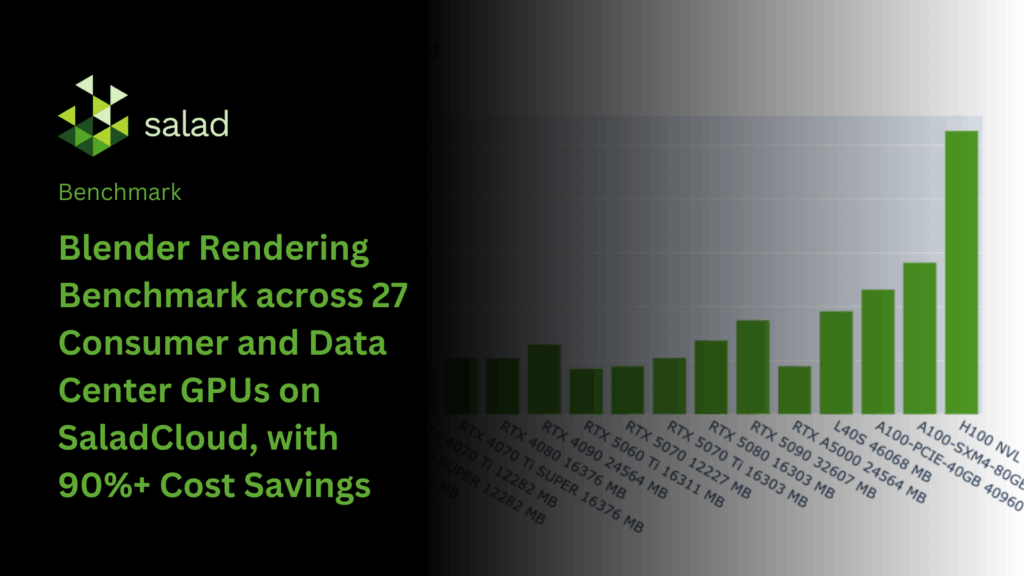
Blender is one of the most widely adopted open-source 3D creation suites, equipped with industry-grade tools for modeling, animation, and rendering. Its Cycles and Eevee engines deliver highly realistic visuals while taking advantage of modern GPU acceleration for significant performance gains. In this analysis, we benchmark Blender rendering on SaladCloud, comparing rendering speed and cost-efficiency across multiple GPU types. The results highlight performance trade-offs between consumer-grade and data center hardware, showing how creative professionals and studios can build scalable, high-performance rendering pipelines on SaladCloud. This approach enables virtually unlimited rendering throughput while reducing costs by over 90% compared to traditional data center GPU solutions. Benchmark Methodology The Blender Benchmark Launcher – Linux CLI is part of the official Blender Open Data Benchmark project. It’s a command-line tool that runs standardized Cycles rendering benchmarks without opening the Blender UI or manually configuring scenes. In Blender’s Cycles engine, a sample represents one light-ray calculation per pixel. At a resolution of 1920×1080, a single frame contains over two million pixels, so increasing the number of samples per pixel produces cleaner, less noisy renders. The benchmark measures how many samples can be completed for all pixels in a frame within a fixed time, providing a consistent metric of machine performance. The benchmark uses three common scenes—Classroom (1920×1080, 300 samples per pixel), Monster (1024×1024, 256 samples per pixel), and Junkshop (2000×1000, 240 samples per pixel)—rendering a single frame from each for a fixed duration (30 seconds) or until the scene’s target samples are reached. Rendering is performed using the Cycles engine on either the CPU or GPU. The total number of samples completed per minute across all scenes is calculated and summed to produce the final score for each test machine. When using NVIDIA GPUs, the Cycles engine supports two rendering backends: CUDA and OptiX. OptiX, built on top of CUDA, leverages hardware RT Cores for accelerated ray tracing. Since not all NVIDIA GPUs have RT Cores, the benchmark only uses the CUDA backend for compatibility, which may result in lower performance. The following command runs the benchmark in the target environment: The Blender Open Data Benchmark score is a relative performance metric based on the total number of samples rendered per minute across the benchmark scenes. It allows comparison across different CPUs and GPUs, but does not directly reflect absolute rendering times or costs for real-world scenes, which depend on factors such as scene complexity, resolution, sample count and specific hardware. To address this, we perform additional benchmarks to measure the actual time required to reach the target samples for a single frame in each scene. From this, we calculate metrics such as average hourly rendered frames and frames per dollar across the combined scenes on a SaladCloud node. The benchmark can be executed using the following command: The rendering process includes scene loading, warm-up, and rendering, with multiple frames in an animation sharing the preparation time, improving rendering performance and cost efficiency per frame. The Blender Open Data Benchmark score already excludes this preparation time. For our additional benchmarks, however, we treat each frame as an independent job, measuring the full end-to-end time. This represents a conservative, worst-case scenario. Please refer to the link for the Dockerfile and benchmark code. When run on SaladCloud, the workflow first executes the benchmarking code, reports the results to an AWS DynamoDB table, and then exits. The data is subsequently downloaded and analyzed using Pandas in JupyterLab. The three benchmark scenes—Classroom, Monster, and Junkshop—each require between 1 and 5 GB of VRAM to render. We benchmark both consumer GPUs (16 vCPUs, 16 GB RAM, all GPU models with ≥ 8 GB VRAM) and data center GPUs (32 vCPUs, 256 GB RAM, all GPU models). On multi-GPU nodes, we set CUDA_VISIBLE_DEVICES to ‘0’ so that only the GPU 0 is accessible to Blender during benchmarking. In total, we gathered data from more than 3,000 Salad nodes spanning 27 GPU models, ensuring broad and representative coverage for each. Batch priority was used to run the benchmarking workloads on SaladCloud, meaning that they could be preempted by high-priority workloads but significantly reduce costs for batch rendering. For pricing calculations on multi-GPU nodes, the cost of CPU and RAM is prorated based on the usage of a single GPU. The hourly cost for each GPU instance type included in the benchmark is shown below: It is worth mentioning that performance can be influenced by many factors, such as operating systems (Windows, Linux, or WSL) and their versions, CPU models, GPU models, and driver versions, CUDA framework versions, Blender versions and additional features enabled in the runtime environment. It is very common to see different results between our benchmarks and those of others. Benchmark Results Key Observations The Blender Open Data Score highlights the significant performance gap between a 16-vCPU setup and various GPUs Blender Cycles with the CUDA backend primarily relies on CUDA cores and performs most computations in single precision (FP32). Rendering performance scales with both the number of CUDA cores and the available memory bandwidth, which is why high-end GPUs consistently outperform lower-end models. However, the flagship cards of an older generation often remain faster than the entry-level models of the next generation, since raw FP32 throughput and bandwidth typically outweigh incremental architectural improvements. Blender Cycles using the CPU backend with 16 vCPUs is much slower than any GPU with the CUDA backend, achieving only a few percent of GPU performance. This is because CPUs have far fewer cores and lower memory bandwidth. Since rendering in Cycles is highly parallel, GPUs—with thousands of FP32 cores optimized for throughput—can complete tasks several to tens of times faster than CPUs. Consumer GPUs, such as the RTX 4090 and 5090, often outperform data center GPUs like the A100 and H100 for Blender Cycles rendering. They are optimized for high FP32 throughput, faster clock speeds, and graphics-focused memory architectures, whereas data center GPUs are designed primarily for FP64 calculations and AI workloads. As a result, these GPUs deliver superior speed and efficiency
Introducing Salad’s Transcription API: superior accuracy now available at 40% of the cost of other providers

Revolutionizing AI Batch Transcription for Enterprises In a world where AI transcription is rapidly becoming a cornerstone for industries like media, customer service, legal, healthcare, and education, accuracy and affordability remain elusive. Most transcription APIs promise high accuracy but come with steep price tags, making large-scale batch transcription impractical for most companies. What’s more egregious is that many companies are left overpaying for lesser accuracy. API prices range between $0.26 to an eye-popping $1.44 per hour for just-par accuracy while charging more for additional features. At Salad, we’ve fixed this. We’re excited to announce the launch of the Salad Transcription API, setting a new industry benchmark for AI batch transcription by combining the highest accuracy rate with the lowest cost per hour. Built for scale and enterprise use cases, this API offers transcriptions, translations, summaries, insights, custom prompts and more for a single price. Why Transcription accuracy and cost matter more than ever Today’s businesses rely on AI Transcription APIs for a wide range of tasks, including: Call Center Analytics: Capturing and analyzing customer interactions. Conversation Intelligence: Transcribing & summarizing calls/meetings to get actionable business insights Media & Entertainment: Creating transcripts, subtitles and captions for audio/video content Medical Documentation: Converting patient consultations into structured records E-learning Platforms: Automating transcription for online courses and content Legal Transcription: Processing and archiving court proceedings. However, the status quo comes with a steep price. AI transcription providers often charge exorbitant fees to offset their R&D costs, model training, and datacenter GPU expenses. For companies requiring millions of hours of audio transcription, these costs become unsustainable. Salad Technologies has broken this cycle. Our new API empowers enterprises to transcribe massive volumes of audio at a fraction of the cost, all while maintaining industry-best accuracy. What Makes the Salad Transcription API Different? 1. No.1 in accuracy – Benchmark-verified Salad Transcription API achieved a 95.1% Word Accuracy Rate (WAR) in English in our accuracy benchmark, outperforming all major market competitors. Here’s how we compare: Deepgram: 3.1% more accurate. 38.4% less cost. Assembly AI: 1.7% more accurate. 56% less cost. Amazon Transcribe: 5.4% more accurate. 89% less cost. Google STT: 4.3% more accurate. 83% less cost. Azure Batch: 3.9% more accurate. 55% less cost. We didn’t stop there. Our API also set record-breaking accuracy rates across multiple languages, including: Spanish: 96.8% WAR German: 96.3% WAR Russian: 96.3% WAR Italian: 93.3% WAR Portuguese: 92% WAR French: 92% WAR These benchmark results, conducted over the CommonVoice 5.1 dataset with over 1 million audio files and 4,500 hours of content, validate Salad’s position as the market leader in transcription accuracy. Thinking to run on Salad? Meet with our Transcription API team. 2. Lowest cost in the industry – 40% less than competitors Cost is a critical consideration for enterprises that require high-volume, batch transcription. For example, contact centers could transcribe upwards of 10 Million hours of audio per month. While most APIs price their services between $0.26 and $1.44 per hour, Salad’s Transcription API is priced at an unbeatable $0.16 per hour – a 40% cost reduction compared to leading alternatives. By leveraging our distributed cloud of 450,000+ GPUs and an open-source, multi-step, multi-modal approach, we’ve drastically reduced the operational costs of transcription and the features on top of it. Unlike other providers that pass the cost of model training and expensive datacenter GPUs onto their customers, we utilize a unique infrastructure that allows us to offer premium accuracy at a fraction of the price. 3. Built for scale – Millions of hours in parallel Batch transcription at scale has traditionally been a bottleneck for enterprises. With asynchronous processing on 1000s of GPUs, Salad Transcription API can handle millions of hours of audio in parallel – making it genuinely built for high-volume enterprise workloads. This level of scalability unlocks new use cases where transcription at scale was previously too expensive or time-consuming: Media archives transcribing thousands of hours of video content. Large-scale call center analytics generating insights from millions of calls. E-learning platforms providing automated transcription for massive course libraries. 4. Unified API with No Hidden Fees Most transcription APIs offer transcription as a core service but charge additional fees for advanced features like: Translation Summarization Speaker diarization LLM-based analysis Custom prompts and vocabulary Salad eliminates complexity and upcharges by bundling all these advanced features under a single, all-inclusive rate. With no hidden fees and no secondary API calls, enterprises get access to a fully unified solution at one predictable price. 5. An ultra low-cost, faster version We are also introducing Salad Transcription Lite for ultra-fast, cheaper transcription for closer-to-real-time needs. This lite version, priced at just $0.03 per hour, will save thousands of dollars for customers looking for reduced latency. How Does Salad Achieve Superior Accuracy at Lower Cost? Open-Source, Multimodal AI Model Unlike competitors that invest heavily in proprietary models (leading to high costs), Salad builds on open-source AI — using transcription models in their most accurate configuration, optimized for long-form transcription. Instead of sacrificing quality for speed, we run a slower but more precise transcription method that takes advantage of high-VRAM GPUs. This approach: Combines state-of-the-art ASR (Automatic Speech Recognition) with advanced NLP. Enhances consistency and accuracy with a sliding window approach that maintains context across audio segments. Improves timestamp precision using language-specific forced alignment models. Delivers high-quality translations and insights using one of the best open-source LLM models available. By fine-tuning these models and running them on Salad’s distributed cloud of GPUs, we achieve benchmark-leading accuracy at a fraction of the typical cost. Why Enterprises Are Switching to Salad Transcription API Unparalleled accuracy across languages With verified benchmarks showing best-in-class WAR and WER (Word Error Rate) across multiple languages, Salad’s API empowers global enterprises to expand into new markets with confidence. Cost savings that unlock new possibilities By cutting transcription costs by 40% or more, companies can: Scale transcription across more departments and projects. Reallocate savings to other AI initiatives or operational growth. Unlock use cases that were previously cost-prohibitive. Frictionless Enterprise Integration Salad Transcription API
Salad Transcription API Accuracy Benchmark: How it outperforms Deepgram, Assembly AI & AWS

Benchmarking Salad Transcription APIs: Salad Transcription and Transcription Lite We recently completed extensive accuracy benchmarks comparing our two transcription APIs – Salad Transcription API and Transcription Lite. Our goal was to measure and compare their accuracy across multiple languages using widely recognized, publicly available datasets and also compare their accuracy against existing transcription solutions. In this blog, we break down the methodology, workflow and results from our Transcription accuracy benchmark. For users interested in recreating the benchmark, we also provide publicly available scripts to recreate the benchmark and test the accuracy results. Overview of our AI Transcription APIs At Salad, we provide two AI transcription APIs offering different features and capabilities to the market. Our two main APIs for Speech-to-text transcription are: For more information about all the features, check out our documentation. Transcription accuracy benchmarking methodology Accuracy is often the most critical factor when evaluating transcription services, particularly for professional applications. To fairly assess our services, we adopted a benchmarking approach similar to what AssemblyAI used, utilizing publicly available datasets. We initially focused on English-language datasets already processed by Assembly AI to get direct comparisons to their results. Datasets Used We selected three datasets for our benchmarks: Workflow Our benchmarking process included: Audio Preprocessing: Audio samples were uploaded to Salad S4 storage. Transcription: Audio files were transcribed using both the Salad Transcription API and Transcription Lite. Normalization: Both the predicted transcripts and the ground truth were normalized using the open-source Whisper Normalizer to ensure consistency by standardizing punctuation, capitalization, and formatting. Normalization ensures that minor formatting differences do not affect accuracy results. Below are examples of how transcripts were adjusted: Original: After Normalization: Original: After Normalization: Accuracy Evaluation: We calculated Word Error Rate (WER) for each file, using the JiWER library, to objectively compare transcription accuracy across datasets. The average WER was then determined for each dataset. You can find all our benchmarking scripts here: https://github.com/SaladTechnologies/salad-transcription-accuracy-benchmarks Here is an example script: Benchmark results: Word Error Rate (WER) for English Dataset Salad Transcription API Salad Transcription Lite API AssemblyAI Universal Amazon Transcribe Google Latest-long Microsoft Azure Batch v3.1 Deepgram Nova 2 OpenAI Whisper CommonVoice 4.90% 18.70% 6.67% 8.98% 17.59% 7.81% 12.43% 8.83% Meanwhile 4.30% 16.70% 4.77% 7.27% 11.67% 6.73% 5.56% 9.75% TED-LIUM 4.20% 8.20% 7.21% 9.12% 11.69% 9.27% 8.98% 7.30% Salad’s Transcription API is cost-effective and accurate. Meet with our transcription team today. Our benchmarks show that the Salad Transcription API consistently delivers the best accuracy in the market compared to other transcription services in the market. Expanding our benchmarks to more languages After comparing our transcription APIs against all major competitors, we expanded our benchmarking efforts to include additional datasets and languages. Our goal is to measure performance across all languages and identify areas for further improvement. The following table presents our latest benchmark results, showing accuracy and Word Error Rate (WER) for Salad Transcription API and Transcription Lite across multiple languages. Dataset Sub-dataset Language Full API Accuracy Lite Accuracy Full API WER Lite WER TED-LIUM tedlium English 95.8% 91.8% 4.2% 8.2% Meanwhile Meanwhile English 95.7% 83.3% 4.3% 16.7% CommonVoice cv-corpus-5.1-2020-06-22 English 95.1% 81.3% 4.9% 18.7% CommonVoice cv-corpus-20.0-delta-2024-12-06 English 93.1% 78.1% 6.9% 21.9% CommonVoice cv-corpus-8.0-2022-01-19 Portugese 92% 55% 8% 45% CommonVoice cv-corpus-10.0-delta-2022-07-04 French 92% 54.3% 8% 45.7% CommonVoice cv-corpus-12.0-delta-2022-12-07 Spanish 94% 58.2% 6% 42.8% CommonVoice cv-corpus-14.0-delta-2023-06-23 Spanish 96.8% 79.5% 3.2% 20.5% CommonVoice cv-corpus-16.1-delta-2023-12-06 Spanish 95.7% 70.9% 4.3% 29.1% CommonVoice cv-corpus-13.0-delta-2023-03-09 German 96.3% 71.1% 3.7% 28.9% CommonVoice cv-corpus-20.0-2024-12-06 Hindi 84% 0% (translates to Eng) 16% 100% CommonVoice Italian 93.3% 54% 6.7% 46% CommonVoice Russian 96.4% 60% 3.6% 40% CommonVoice cv-corpus-17.0-2024-03-15 Hebrew 84.2% 12% 15.8% 88% CommonVoice cv-corpus-19.0-2024-09-13 Kazakh 51% 0% 49% 100% CommonVoice cv-corpus-9.0-2022-04-27 Urdu 78.8% 8.3% 21.2% 91.7% Salad Transcription API performs exceptionally well in English and major European languages, achieving high accuracy in: English, Spanish, German, French, Portuguese, Italian, and Russian. However, there is room for improvement in certain languages, particularly in: Thai, Kazakh, Hebrew, Hindi and Urdu. Transcription Lite currently performs well in English as the base language, as it’s optimized for speed. Industry-Leading Pricing While accuracy is a very important factor in choosing a transcription service, cost is just as important especially for large-scale applications. Salad’s Transcription APIs are not only among the most accurate but also the most affordable APIs compared to competitors. Pricing Breakdown This makes Salad Transcription API the cheapest high-accuracy solution on the market, and Transcription Lite one of the most cost-effective, close to real-time transcription services available. Key Takeaways from Our Benchmarks Our benchmarking process, comparing Salad Transcription API and Salad Transcription Lite against major transcription providers and across multiple languages, has revealed several insights: 1. Leading accuracy in Transcription 2. Challenges in low-resource languages 3. Transcription Lite accuracy vs speed Next Steps Expanding our dataset coverage to include more datasets and languages. Improving transcription for non-English languages, particularly in low-resource languages. We will continue updating our benchmarks and improving our transcription models to provide the best value, accuracy, and performance in the market. Stay tuned for more updates! Schedule a call with our expert transcription team. SaladCloudSaladCloud is the world’s largest distributed cloud computing network with 11,000+ daily GPUs and 450,000 GPUs contributing compute, all at the lowest cost in the market.
There’s An AI For That scales user-generated AI tools with SaladCloud at 50% less cost

Fast Facts about TAAFT & SaladCloud: The world’s largest AI tools platform launches AI-powered user apps In late 2022, well before ChatGPT brought AI to the masses, Andrei, a developer and marketer, quietly launched There’s An AI For That (TAAFT) – a directory of AI tools. Within its first week, the site saw over 100,000 organic visitors. No ads. No marketing blitz. Zero dollars spent in advertising. Today, a team of 15 have grown TAAFT into the world’s largest AI tools platform, with 5 Million active users every month. As more users and developers joined the TAAFT community, they wanted to let their community build AI tools and launch them. Enter ‘Mini Tools’ – From text generators to image tools, from content creation to data analysis, users are now creating and deploying thousands of AI-powered “Mini Tools” directly on TAAFT. The rapid growth of Mini Tools presented a new challenge: how do you power a sprawling ecosystem of AI tools at scale for a reasonable cost? This is where TAAFT turned to SaladCloud. The search for scalable compute When TAAFT began planning the debut of their Mini Tools, one of their first priorities was finding an infrastructure partner that could support the rapid growth of these user-generated AI tools. The team explored their options, sifting through hyperscalers and neoclouds alike. Eventually, SaladCloud stood out. “We didn’t want a one-size-fits-all solution. SaladCloud’s custom deployment options gave us the control we needed to optimize each AI tool for peak performance and efficiency, ensuring a seamless user experience. Their price-to-performance ratio was unmatched,” said Andrei. Andrei, Founder of TAAFT GPU utilization and custom model deployment To meet the computational demands of Mini Tools, SaladCloud’s infrastructure was utilized to deploy high-end Nvidia GPUs for compute-intensive models, ensuring high-performance processing for tasks such as image generation and complex data analysis. For high-throughput, cost-effective inference endpoints, lower-end GPUs were used, balancing performance with affordability. SaladCloud’s custom model deployment capabilities allowed for tailored processes, ensuring optimal performance and resource utilization across the diverse functionalities of Mini Tools. “The beauty of our platform is its diversity. We need to support everything from lightweight text tools to incredibly demanding image generation models. SaladCloud’s flexibility, allowing us to leverage a variety of GPUs and scale easily, is critical” Reliable, easy user experience With SaladCloud, TAAFT has deployed user-generated AI apps across multiple modalities. One example is their speech-to-text service, powered by Salad Transcription API, which will enable TAAFT users to transcribe audio – including YouTube videos – directly into usable text, using optimized batch inference pipelines. Beyond cost-efficiency, the smooth user experience and platform reliability were other key benefits of SaladCloud. “A smooth, reliable experience is paramount for our users. SaladCloud’s intuitive platform and robust API have minimized downtime and allowed us to focus on delivering value, not troubleshooting infrastructure” Scaling smart with big savings While final cost savings data is still in progress, early results suggest SaladCloud is on track to reduce TAAFT’s infrastructure costs by at least 50% compared to other providers. “We’re still gathering precise data, but early indications suggest SaladCloud could deliver a 50% reduction in our infrastructure costs. That’s a significant number, and it allows us to focus on what we do best: curating and promoting incredible AI.“ The Bigger Picture: Democratizing AI Creation At its core, TAAFT is about more than just AI tools. It’s about empowering creators – whether they’re hobbyists or startups – to build AI-driven applications quickly and easily. “Our goal with There’s An AI For That is to democratize access to powerful AI tools. Making those tools available is only half the battle – they need to be affordable and reliable too, and that’s where SaladCloud has been a game-changer.” Andrei and the team believe in a future where AI tool creation will be as ubiquitous as social media profiles came about a decade ago. “The future of AI isn’t just about building better models; it’s about making those models accessible to everyone. SaladCloud is playing a vital role in making that future a reality” SaladCloudSaladCloud is the world’s largest distributed cloud computing network with 11,000+ daily GPUs and 450,000 GPUs contributing compute, all at the lowest cost in the market.
SaladCloud becomes the first to offer NVIDIA RTX 5090/5080s, thanks to the power of community

SaladCloud offers RTX 5090/5080 just 24 hours after launch On January 30th, 2025, Jensen Huang (and his customary leather jacket) launched the NVIDIA RTX 5000 series GPUs at CES. For the GPU-hungry AI/ML crowd, the 5000 series GPUs – RTX 5090, 5080, 5070 Ti and 5070, offered benefits similar to the earlier RTX/GTX GPUs – a viable, affordable alternative to the more expensive datacenter GPUs for many workloads. Thanks to the power of our crowdsourced model and community, SaladCloud became the first provider to offer the latest GPUs from Nvidia’s stable for compute workloads. Just 24 hours after the launch, our internal Slack channel beeped with the message: “We have the first 3 5090s enabled on the network” One of our “chefs” – compute providers on our distributed network – from Italy offered their RTX 5090 on SaladCloud. Soon two other RTX 5090s followed – from the US and the UK. By the end of the week, dozens of RTX 5090s were live on our distributed cloud platform, ready to power cutting-edge AI/ML, video and rendering workloads before any major cloud provider could even list them. How did we pull this off? The answer lies in our unique community cloud model. How we brought RTX 5090s to the cloud first Traditional cloud providers source GPUs through centralized data centers, which means they’re at the mercy of long lead times and supplier bottlenecks. For users, this often translates into two things: SaladCloud takes a different approach. We utilize the power of a global community of individual GPU owners who contribute their latent hardware time to the network in exchange for rewards. There are more than 100 Million AI-enabled GPUs in the world today. A majority of them sit idle for 18-20 hrs a day. This unused compute is what we harness to power SaladCloud. That means when NVIDIA’s latest RTX 5090 GPUs started hitting the market, SaladCloud was ready to integrate them immediately. Individual power users, AI enthusiasts, and institutions with early access to these GPUs began contributing their compute to SaladCloud, allowing us to offer RTX 5090s to developers before anyone else. This also means that when AI/ML companies need to scale to 100s of GPUs and can’t find supply on hyperscalers and other clouds, SaladCloud can tap into our network of 450,000+ GPU owners to bring more GPUs online in hours. While many companies are working on a distributed cloud, SaladCloud is the largest distributed cloud in the world with 20,000+ daily active GPUs and a pool of 450,000+ GPUs to activate from. Cost of 5090/5080 on SaladCloud and how to deploy them The RTX 5090 is now available from just $0.27 per hour. The RTX 5080 costs just $0.195 per hour with batch priority. These are the lowest prices in the market today to rent these GPUs. To deploy these GPUs for your workloads, head over to portal.salad.com, create an account and start renting. What RTX 5090/5080 means for AI developers The RTX 5090 represents a massive leap in GPU performance. With 32GB of VRAM, a higher core count, and significantly increased memory bandwidth, it’s an AI powerhouse designed to handle the most demanding workloads. Puget Systems has already benchmarked both 5090 and 5080 for many use cases. Whether you’re deploying real-time inference at scale, running generative AI workloads, or rendering 3D models and videos, the 5090 is a good upgrade for many AI use cases. For example, Flux.1 fine-tuning for AI image generation will get better cost-performance on a 5090 on SaladCloud. The 5080 also shows robust performance for many AI use cases – as good as the 4080 for image generation (SD.Next), for example. Key benefits of running on SaladCloud: Why the future of Cloud Compute is community powered SaladCloud is proving that a decentralized cloud can beat the hyperscalers and datacenter GPUs when it comes to speed, scalability, and cost. By tapping into the latent power of consumer GPUs sitting idle around the world, we’ve built a cloud that scales dynamically with market demand while saving thousands in compute cost. The result? Developers and AI teams get faster, lower-cost access to cutting-edge GPUs like the 5090, at a fraction of the cost of traditional cloud services. If you’re ready to experience the next generation of AI compute, start deploying workloads on RTX 5090s today. 🚀 Sign up and start building now SaladCloudSaladCloud is the world’s largest distributed cloud computing network with 11,000+ daily GPUs and 450,000 GPUs contributing compute, all at the lowest cost in the market.
DeepSeek TGI Benchmark: Build highly scalable and cost-effective LLM applications on SaladCloud, at 70% less cost than hyperscalers

DeepSeek LLM benchmark on SaladCloud LLM applications need a highly scalable and cost-effective GPU infrastructure to meet rapidly evolving demands. While the data center GPUs, such as A100 and H100, from major cloud providers offer high throughput and exceptional performance, they are often too costly and excessive for many use cases, such as text classification, translation, summarization, and personalization. Additionally, some cloud providers emphasize committed resource usage, which doesn’t align with the flexible business models of startups and other organizations. SaladCloud, leveraging tens of thousands of consumer GPUs, provides fine-grained, diverse resource options with unmatched scalability, flexibility and cost-effectiveness. We’ve seen an increasing number of customers successfully running and fine-tuning their LLM models on SaladCloud, achieving over 70% cost savings compared to hyperscalers. In this article, we share results from a DeepSeek TGI benchmark on SaladCloud, showing how companies can develop highly scalable and cost-effective LLM applications. Interested in deploying on SaladCloud? Contact our support team today. DeepSeek R1 is a new, highly powerful open-source reasoning model with performance on par with OpenAI’s o1. The distilled and fine-tuned smaller dense models by using the reasoning data generated by DeepSeek R1, also deliver exceptional results on benchmarks. Hugging Face’s TGI v3 is the latest version of its toolkit for deploying and serving LLMs, offering significant performance enhancements with zero configuration required. In this comprehensive benchmark, we explore the deployment and performance evaluation of a typical LLM inference system using Hugging Face’s TGIv3 and DeepSeek-R1-Distill-Llama-8B on SaladCloud. Additionally, we share best practices from our customers for building high-throughput, reliable and cost-effective LLM applications. All code and configurations are available in this Github repository. The DeepSeek benchmark image The official Hugging Face’s TGI image can be deployed on SaladCloud directly, without any modifications. However, to fully leverage the distributed and dynamic nature of SaladCloud, we recommend building a custom wrapper image with the following features for production: Providing input prompts and streaming the generated tokens on SaladCloud There are two primary ways for providing input prompts to your LLM models and streaming the generated tokens on SaladCloud: Deploying SaladCloud’s Container Gateway is the quickest approach. The TGI server on instances should listen on an IPv6 port, and the gateway can map a public URL to this port, forwarding multiple client requests to instances concurrently. Inference time can vary significantly with different prompts. So configuring the Least Number of Connections algorithm is crucial to manage these fluctuations effectively. If client applications send more requests than the TGI server can handle, excessive resource usage may lead to errors and request timeouts. To improve system robustness, the TGI server can proactively reject excessive requests as a backpressure mechanism, while client applications can implement traffic control to stop accepting new requests from users during periods of congestion. A few customers have successfully implemented a Redis-based near-real-time queue on SaladCloud, supporting regional deployment and being platform-independent. In this setup, client applications send requests to a Redis cluster, while a Redis worker, included in the image, pulls jobs (input prompts) from the cluster, invokes the TGI server, and streams the generated tokens. This queue-based approach may enhance resilience against traffic spikes and variations in inference time, as instances only fetch new jobs after completing the current ones. In this benchmark, the container gateway will be deployed to manage access. Please refer to the Dockerfile for the benchmark image, which is built on the official TGI image. This image includes testing tools and performs an initial check (VRAM, CUDA version) before starting the TGI server. Deployment to SaladCloud Currently, all container instances, regardless of location, are centrally accessed through SaladCloud’s Container Gateway in the U.S. To reduce latency, we can use the SaladCloud Python SDK to deploy container groups specifically within the U.S. The example code creates two separate container groups, one for the RTX 3090 and the other for the RTX 4090, each group consisting of 5 replicas. Using the container gateway for instances in other regions may introduce additional latency, typically in the range of several hundred milliseconds. This is generally acceptable since LLM inference usually takes much longer (tens of seconds). However, for latency-sensitive applications that require local access, a Redis-based queue should be considered. For more information on deploying LLMs on SaladCloud, please refer to this guide. Benchmarking tool and methodology We use the open-source LLMPerf for load testing, which allows testing over public endpoints by sending concurrent requests with varying prompt lengths through OpenAI-compatible APIs. It measures key metrics such as Time-to-First-Token (s), End-to-End Latency (s), Request-Output Throughput (tokens/s), and more. Additionally, there is a performance leaderboard for several API providers across various LLM models, benchmarked using LLMPerf. Each LLM model uses its own tokenizer, meaning the same prompt may result in different token counts across models. To maintain consistency and comparability, LLMPerf standardizes token counting by using the LlamaTokenizer for both input and output tokens, regardless of the model being tested. For more information, refer to the additional details about the benchmarking samples generated by LLMPerf. We have updated the benchmark code to include additional metrics based on SaladCloud pricing, such as infrastructure cost, cost per 1K requests and cost per 1M output tokens. For this benchmarking test, we send concurrent requests to the two container groups (the RTX 3090 and the RTX 4090), varying the number of concurrent requests and combinations of prompt and output lengths, and then collect the results. Metrics like TTFT and E2E Latency are also affected by the distance from the gateway to the client location, which, in this test, is California. DeepSeek benchmark results and observations TGI utilizes a continuous batching algorithm, dynamically adding requests to the running batch for optimal performance. As the number of concurrent requests increases from 5 to 10, 20, 40, and 80 for both the two 5-replica container groups, the total output throughput improves by over 500% while the cost per 1M output tokens drops by more than 80%. However, both per-request output throughput and time degrade as the number of concurrent
1000s of GPUs, over 50% savings: Klyne accelerates AI drug discovery on SaladCloud

Accelerating early-stage drug discovery at low-cost with AI The journey of drug discovery is a winding, often frustrating road defined by risk, long timelines, and staggering costs. A typical pharmaceutical company spends up to $2 billion and more than 15 years from the discovery of a drug target to an approved treatment. The high costs involved in early-stage drug discovery – the process of finding potential drug candidates – are especially frustrating & daunting. This was the spark for Klyne AI. CEO Zachary Lawrence comes with a background in mathematics and computer science. He also led investments within the biotech and drug discovery sectors. The challenges of high-risk investments and slow, expensive drug development cycles led to a crucial realization: early-stage drug discovery needed a technological overhaul. Klyne was born in late 2023 to fix this – to help accelerate early-stage drug discovery at low-cost with AI-powered software that improves the efficiency of finding novel hits and optimizing them. “We help smaller companies and startups go from spending hundreds of thousands of dollars to just tens of thousands of dollars – an order of magnitude difference”, says Zack. Klyne’s USP: Proprietary AI model. Pay-go business model. Klyne’s USP lies in their proprietary AI models, including the SPARC model, designed to predict binding affinity — a key measure of how well a drug will interact with a target protein. Klyne’s machine learning-driven predictions are as accurate as anything currently available. They also demand just 1% of the total compute power compared industry standard approaches. This was made possible by the construction of a large proprietary training dataset. There’s another model in Klyne’s arsenal that gives them an advantage: their business model. Rather than charging high upfront subscription fees, Klyne offers pay-as-you-go services and software licenses, democratizing access to advanced drug discovery tools for startups and small businesses. Companies can provide Klyne with a protein file, and their software will analyze trillions of compounds to deliver a list of chemical structures that are very likely to bind to the protein. Since many companies do not have in silico drug discovery expertise in-house, most companies elect to use their white-glove service to ensure the discovery pipeline is set-up in the most optimal way. The challenge in scaling computational resources for drug discovery However, like many startups in the biotech and life sciences space, Klyne faced significant challenges in scaling their operations, particularly around the high computational costs and burst GPU requirements associated with molecular dynamics (MD) simulations. Zack adds, “We needed massive amounts of low-cost compute resources to run 100s of thousands of molecular dynamics simulations. Even with the most efficient algorithms, these simulations are slow, lengthy, and expensive to run. It’s crucial to have as many GPUs as possible at the cheapest price.” Traditional cloud providers offered expensive, high-performance GPUs – ideal for AI training, but are not cost-effective of molecular dynamics simulations. Klyne’s GPU needs varied from tens to hundreds to thousands, which made scaling on hyperscalers prohibitively expensive and challenging. This is where SaladCloud’s flexible, cost-effective cloud computing services became an essential part of Klyne’s success. “You don’t really want higher-end GPUs for your MD simulations. Other cloud providers are focused on providing high-end GPUs. But molecular dynamics is a different market with different needs. SaladCloud’s lower-end GPUs offer way better cost-efficiency for MD simulations” “We may ramp up to 10,000 simulations and ramp down quickly and often. So renting GPUs wasn’t an option. Spot pricing on GCP/AWS is so much more expensive for molecular dynamics simulations than SaladCloud. Plus they don’t always have availability of 1000s of GPUs. With SaladCloud, we can scale up to 1000s of GPUs quickly saving at least 50% compared to other alternatives.” Zachary Lawrence, CEO of Klyne AI SaladCloud helps Klyne scale fast at a low-cost on consumer GPUs While traditional cloud providers offered high-end GPUs, SaladCloud’s distributed infrastructure is tailored to fit the specific needs of drug discovery simulations – allowing companies like Klyne access to 1000s of consumer GPUs at the lowest market cost. “We may ramp up to 10,000 simulations and ramp down quickly and often. So renting GPUs wasn’t an option. Spot pricing on GCP/AWS is so much more expensive for molecular dynamics simulations than SaladCloud. Plus they don’t always have availability of 1000s of GPUs. With SaladCloud, we can scale up to 1000s of GPUs quickly saving at least 50% compared to other alternatives”, added Zack. “The downside of interruptible instances do exist but can be overcome with specific code to save snapshots”. Whether it’s running thousands of simulations or conducting a more targeted molecular dynamics simulation, Salad’s flexibility has been a game-changer for Klyne’s business. “Salad’s team has been a valuable partner for us, building Kelpie for our batch needs and being really customer friendly. We can directly connect with Salad’s engineering team and get the kind of support we wouldn’t from a bigger cloud provider”, adds Zack. The future of AI drug discovery: Klyne, SaladCloud, and beyond Like any AI startup, Klyne has faced infrastructure challenges along the way. The transition from traditional infrastructure to distributed cloud computing was a learning curve. Salad’s support team worked directly with Klyne to develop best practices for running molecular dynamics simulations at low-cost and high-scale on a distributed cloud. By enabling companies to predict drug affinity early in the process and reduce reliance on costly experimental testing, Klyne is helping to make the entire process more efficient and cost-effective. Their platform is used by a range of clients—from academic researchers at universities to enterprise-level drug discovery teams at large pharma companies. “We’re creating the next wave of drug discovery software,” said the Klyne founder. “And SaladCloud is helping us get there faster.” SaladCloudSaladCloud is the world’s largest distributed cloud computing network with 11,000+ daily GPUs and 450,000 GPUs contributing compute, all at the lowest cost in the market.