
Note: Prices have fallen considerably since this benchmark was conducted, so actual costs will be even lower!
Benchmarking GROMACS for Molecular Simulation on Consumer GPUs
In this deep dive, we will benchmark GROMACS on SaladCloud, analyzing simulation speed and cost-effectiveness across a spectrum of small, medium, and large molecular systems. Additionally, we will provide recommendations for selecting the most appropriate resource types for various workloads on SaladCloud.
Building on the OpenMM benchmark on SaladCloud and our continuous efforts to optimize system architecture and batch job implementation, we have achieved a 90% cost savings by using consumer GPUs for molecular simulations with GROMACS, compared to CPUs and data center GPUs.
This capability enables effective static and dynamic load balancing across the system’s various components.
GROMACS is a highly optimized, open-source software package for molecular dynamics simulations. Researchers in fields like biochemistry, biophysics, and materials science widely use it to study the physical movements of atoms and molecules over time. GROMACS stands out for its exceptional performance compared to other programs, efficiently leveraging both CPU and GPU resources. This capability enables effective static and dynamic load balancing across the system’s various components.
Are you running more than $250K/yr in MDS compute? Migrate to the lowest cost GPU cloud with free, white-glove engineering support.
GROMACS benchmark methodology
The gmx mdrun is the main computational chemistry engine within GROMACS. The following command is to perform molecular dynamics simulations in the target environment:
gmx mdrun -s input_filename.tpr -nb gpu -pme gpu -bonded gpu -update gpu -ntmpi 1 -ntomp 8 -pin on -pinstride 1 -nsteps 200000 -deffnm output_filenameThe mdrun program reads the input TPR file (-s), which contains the initial molecular topology and parameters, and produces several output files (-deffnm) with different extension names for logs, trajectories, structures and energies.
GROMACS relies on close collaboration between the CPU and GPU to achieve optimal performance. Although many calculations can be offloaded to the GPU using the options (-nb, -pme, -bonded, -update), the program still demands considerable CPU processing power and multiple threads for task management, communication, and I/O operations. To fully utilize a powerful GPU, GROMACS also depends on robust CPU performance.
While running more OpenMP threads than the number of physical cores could be beneficial in certain situations for GROMACS, but for our benchmark test, we only selected Salad nodes with CPUs that have 8 or more cores and configured each node to run 8 OpenMP threads (-ntmpi, -ntomp).
We used GROMACS 2024.1 with CUDA 11.8 to build the container image. When running on SaladCloud, it first runs the simulations against typical molecular systems, reports the test data to an AWS DynamoDB table, and then exits. Finally, the data is downloaded and analyzed using Pandas on JupyterLab.
Two key performance indicators are collected and analyzed during the test:
ns/day stands for nanoseconds per day. It measures simulation speed, indicating how many nanoseconds of simulated time can be computed in one day of real time.
ns/dollar stands for nanoseconds per dollar. It measures cost-effectiveness, showing how many nanoseconds of simulated time can be computed for one dollar.
Below are the two scenarios and the methods used to collect data and calculate the final results:
| Scenario | Resource | Simulation Speed (ns/day) | Cost Effectiveness (ns/dollar) |
|---|---|---|---|
| Consumer GPUs | 8 cores for 8 OpenMP threads 30 GPU types | Create a container group with 100 instances with all GPU types on SaladCloud, and run it for a few hours. Once the code execution is finished on an instance, SaladCloud will allocate a new node and continuously run the instance. Collect test data from thousands of unique Salad nodes, ensuring sufficient samples for each GPU type. Calculate the average performance for each GPU type. | Pricing from the SaladCloud Price Calculator: $0.072/hour for 16 vCPUs, 8GB RAM $0.015 ~ $0.18/hour for different GPU types (Priority: Batch ) https://salad.com/pricing |
| Data Center GPUs | 16 Cores for 16 OpenMP threads A40 48GB A100 40GB H100 80GB | Use the test data in the GROMACS benchmarks by NHR@FAU. | The lowest prices are selected from the data center GPU market, that closely match the resource requirements: $1.86/hour for A40 (24 vCPUs) $1.29/hour for A100 (30 vCPUs) $2.99/hour for H100 (30 vCPUs) https://getdeploying.com/reference/cloud-gpu |
It is worth mentioning that performance can be influenced by many factors, such as operating systems (Windows, Linux, or WSL) and their versions, CPU models, GPU models, and driver versions, CUDA framework versions, GROMACS versions, and additional features enabled in the runtime environment. It is very common to see different results between our benchmarks and those of others.
Benchmark Results
Here are six typical biochemical systems used to benchmark GROMACS:
| No | Model Description | Size |
|---|---|---|
| 1 | R-143a in hexane (20,248 atoms) with very high output rate | Small |
| 2 | A short RNA piece with explicit water (31,889 atoms) | Small |
| 3 | A protein inside a membrane surrounded by explicit water (80,289 atoms) | Medium |
| 4 | A protein in explicit water (170,320 atoms) | Medium |
| 5 | A protein membrane channel with explicit water (615,924 atoms) | Large |
| 6 | A huge virus protein (1,066,628 atoms) | Large |
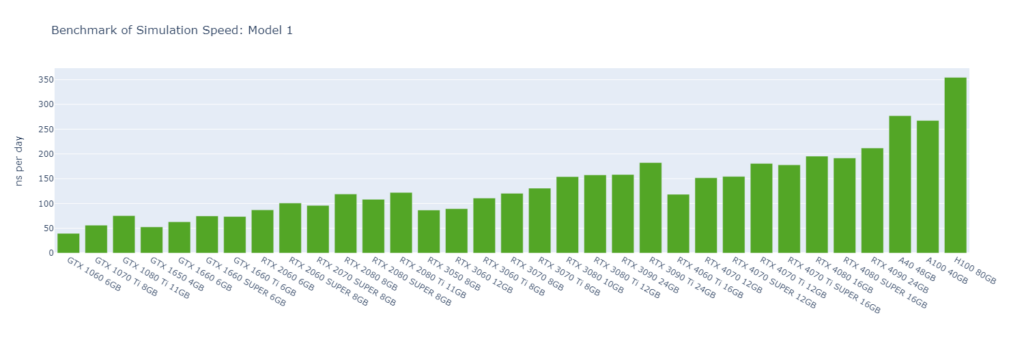
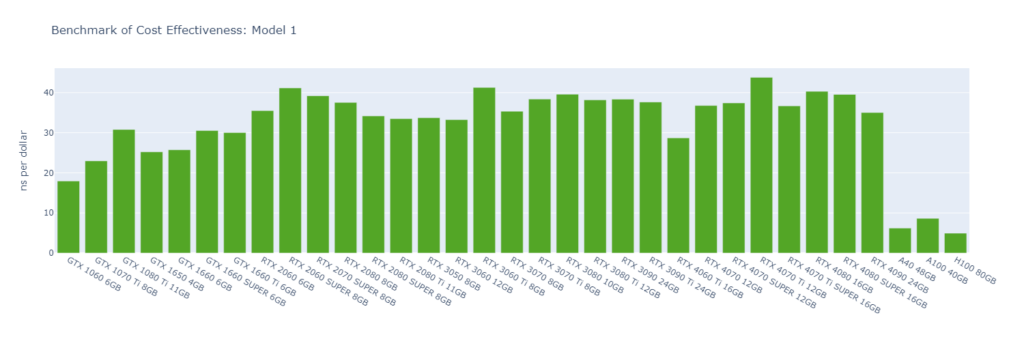
Model 1: R-143a in hexane (20,248 atoms) with very high output rate
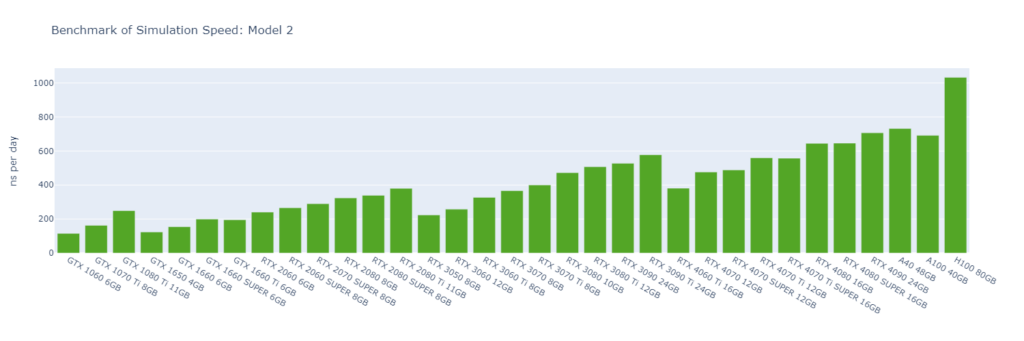
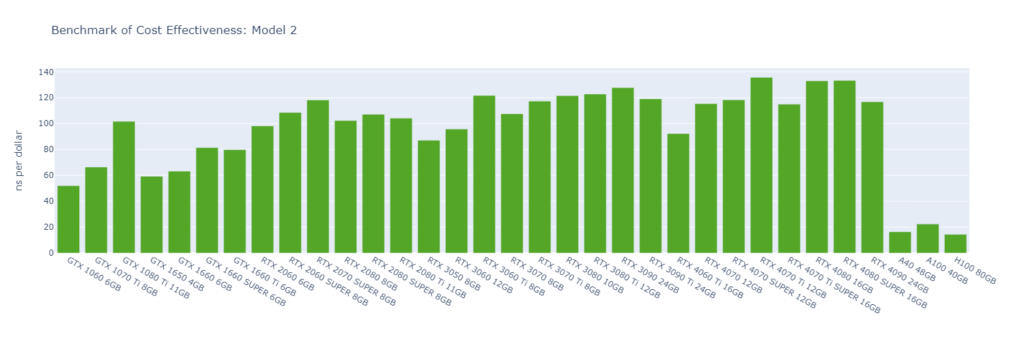
Model 2: A short RNA piece with explicit water (31,889 atoms)
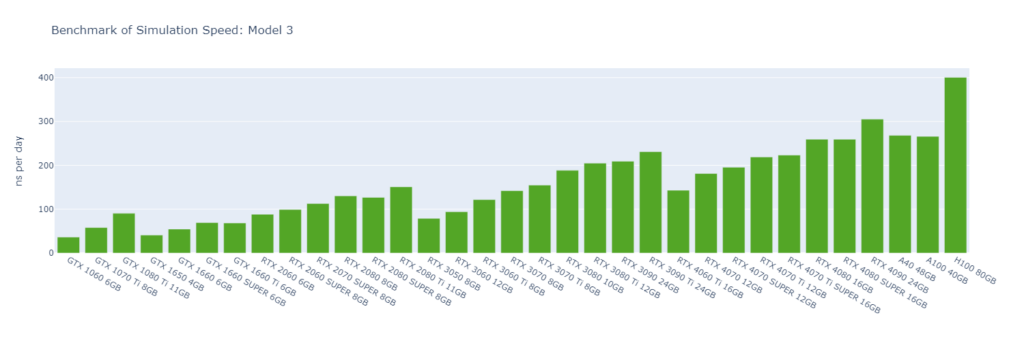
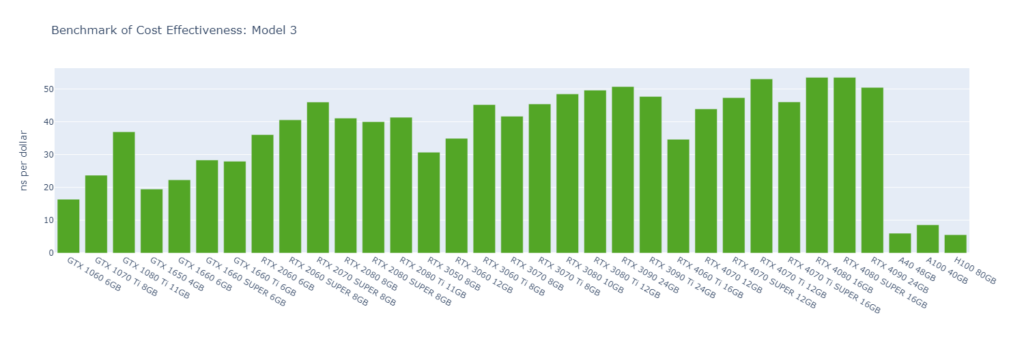
Model 3: A protein inside a membrane surrounded by explicit water (80,289 atoms)
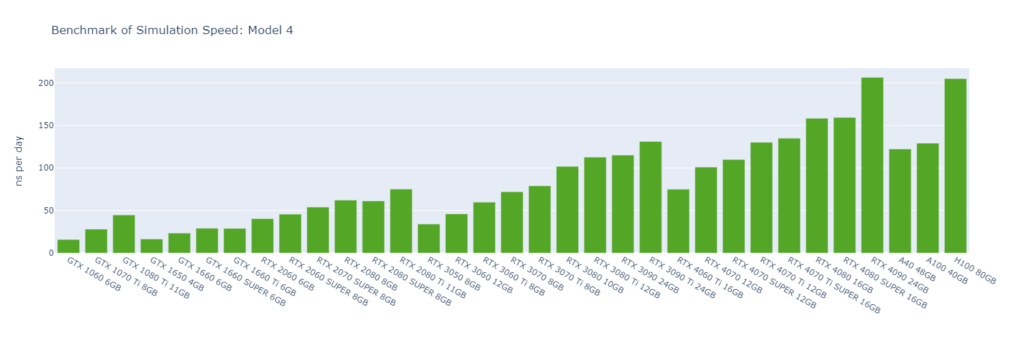
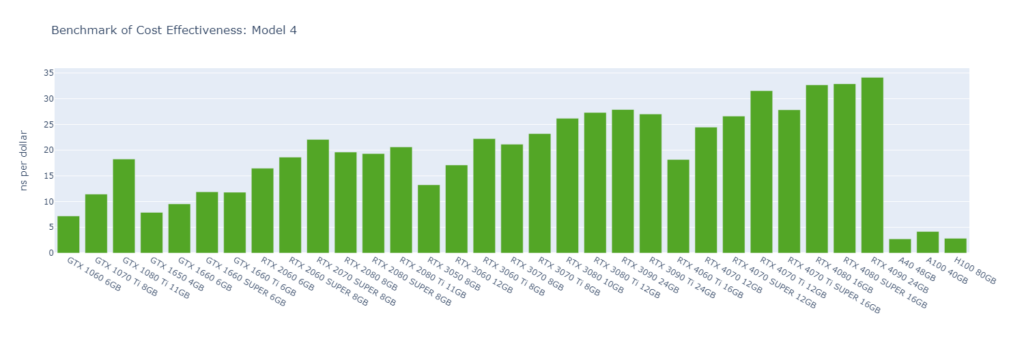
Model 4: A protein in explicit water (170,320 atoms)
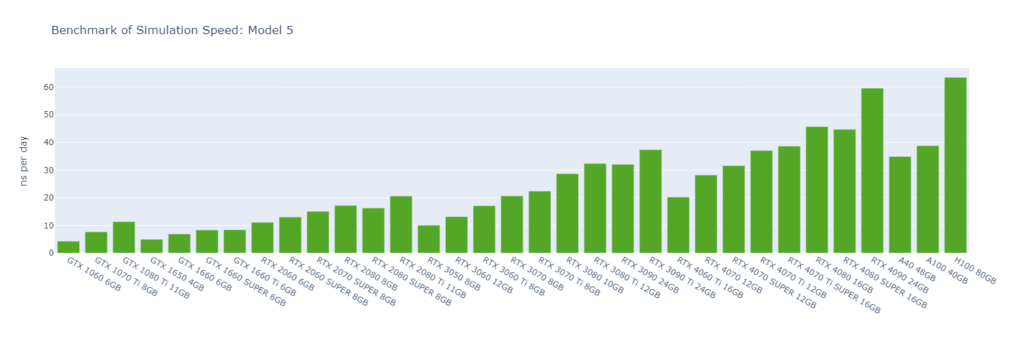
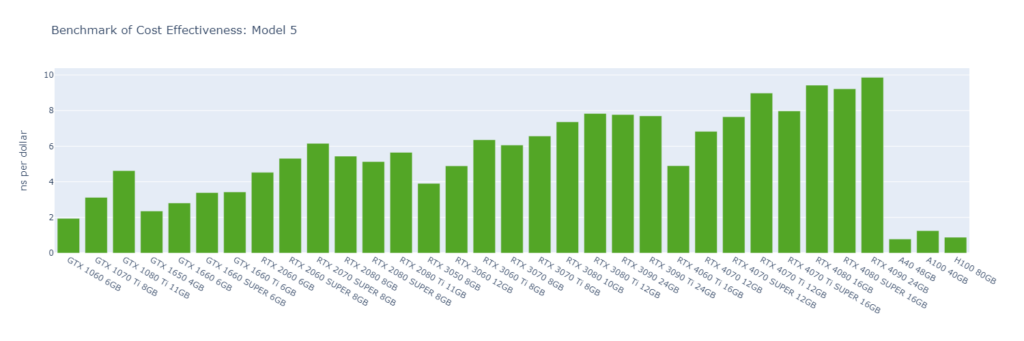
Model 5: A protein membrane channel with explicit water (615,924 atoms)
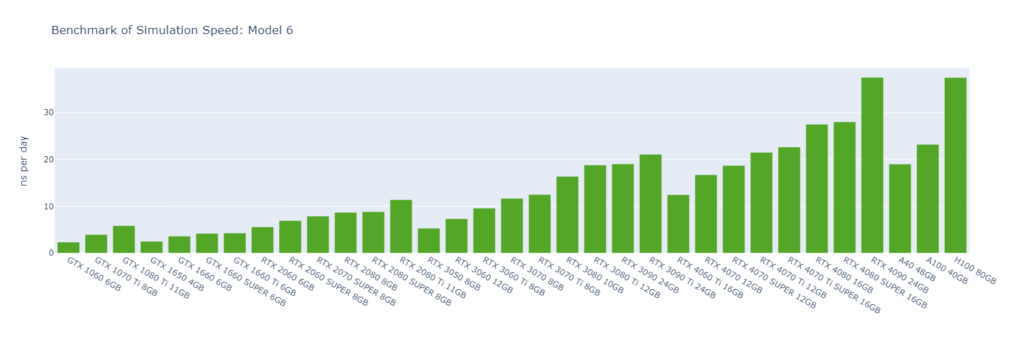
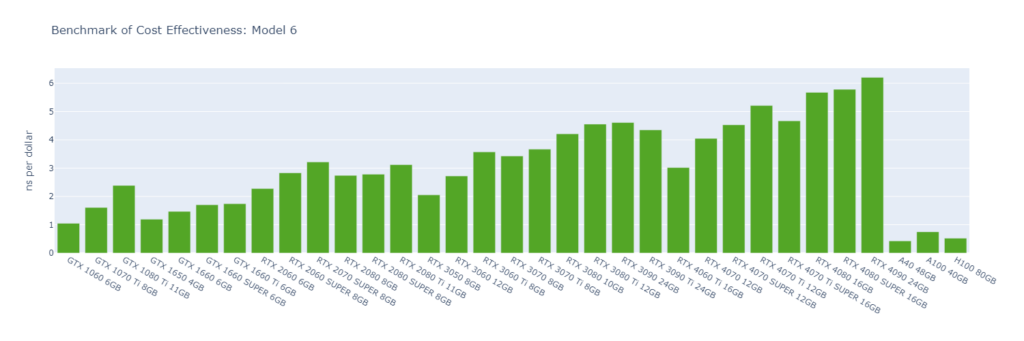
Model 6: A huge virus protein (1,066,628 atoms)
Observations from the GROMACS benchmark
Here are some interesting observations from the GROMACS benchmarks:
The VRAM usage for all simulations is only 1-2 GB, which means nearly all GPU types can theoretically be utilized to run these models.
GROMACS primarily utilizes the CUDA Cores of GPUs (not Tensor Cores), and typically operates in single-precision (FP32). High-end GPUs generally outperform low-end models in simulation speed due to their greater number of CUDA cores and higher memory bandwidth. However, the flagship model of a GPU generation often surpasses the low-end models of the following generation.
For smaller models, GPUs are often underutilized, and communication between the CPU and GPU can become a bottleneck, making CPU performance a critical factor in overall system performance. On nodes with GPUs of similar performance, higher CPU clock speeds and more physical cores usually lead to better performance. Data center GPUs are typically paired with more powerful CPUs that have additional cores, allowing them to run GROMACS significantly faster than consumer GPUs in Models 1 and 2.
Large models can fully exploit the vast number of CUDA Cores and high memory bandwidth offered by high-end GPUs. As models become more complex with additional molecules and atoms, the GPU’s role becomes increasingly crucial, and the performance gap between low-end and high-end GPUs widens. For instance, in Model 1, the H100 outperforms the 1060 by 9x, while in Model 6, the 4090 surpasses the 1060 by 16x.
As models increase in size, consumer GPUs (4080 and 4090) with 8 OpenMP threads begin to match or even surpass the performance of data center GPUs (A40, A100, and H100) with 16 OpenMP threads, particularly from Model 3 onwards due to their higher FP32 TFLOPS.
While molecular simulation jobs require minimal VRAM, this doesn’t necessarily make low-end GPUs the better options for cost savings compared to high-end GPUs. Low-end GPUs may offer competitive pricing for smaller models, but as model sizes increase, high-end GPUs become more cost-effective due to their superior performance.
Across all models, consumer GPUs can be significantly more cost-effective than data center GPUs, with cost savings ranging from 8x in Model 1 to 14x in Model 6. This makes them an ideal choice for batch molecular simulation jobs.
In need of a more cost-effective GPU cloud for simulation scalability? Migrate to SaladCloud, covering up to 100TB of egress fees for limited time.
Recommended Resource Types for GROMACS Workloads on SaladCloud
| Model | Size | Most Cost-Effective GPUs | Best Performing GPUs |
|---|---|---|---|
| Small | < 50,000 atoms | 4070 Ti 3060 Ti 4080 | 4090 4080 SUPER 4080 |
| Medium | 50,000 ~ 500,000 atoms | 4090 4080 4070 | 4090 4080 SUPER 4080 |
| Large | > 500,000 atoms | 4090 4080 4070 | 4090, 4080 SUPER 4080 |
For smaller models or workloads utilizing high-end GPUs, experimenting with a higher number of OpenMP threads than the number of physical cores could further enhance performance.
Given the complexity and variety of these biochemical systems, the best-performing GPU and the most cost-effective GPU for each model might differ. Before deploying your workloads extensively SaladCloud, we recommend conducting a quick benchmarking test to identify the optimal resource types for your models. This will allow you to optimize your workloads for reduced costs, improved performance, or both.
SaladCloud: The Right Choice for Molecular Simulations
Based on benchmark tests with both OpenMM and GROMACS, consumer GPUs are clearly the right choice for running molecular simulation jobs. They offer performance comparable to data center GPUs while delivering over 90% cost savings. With the right design and implementation, we can build a high-throughput, reliable, and cost-effective system on SaladCloud to run simulation jobs at a very large scale.
If your needs involve running millions of simulation jobs quickly or requiring access to hundreds of GPUs or various GPU types within minutes for testing or research purposes, SaladCloud is the ideal platform.
Are you running more than $250K/yr in MDS compute? Migrate to the lowest cost GPU cloud with 1:1 prepaid credit matching from SaladCloud!
SaladCloud is the world’s largest distributed cloud computing network with 11,000+ daily GPUs and 450,000 GPUs contributing compute, all at the lowest cost in the market.


