
Note: Prices have fallen considerably since this benchmark was conducted, so actual costs will be even lower!
Benchmarking OpenMM for Molecular Simulation on consumer GPUs
OpenMM is one of the most popular toolkits for molecular dynamics simulations, renowned for its high performance, flexibility, and extensibility. It enables users to easily incorporate new features, such as novel forces, integration algorithms, and simulation protocols, which can run efficiently on both CPUs and GPUs. This analysis uses typical biochemical systems to benchmark OpenMM on SaladCloud’s network of AI-enabled consumer GPUs. We will analyze simulation speed and cost-effectiveness in each case and discuss how to build high-performance and reliable molecular simulation workloads on SaladCloud. This approach supports unlimited throughput and offers over 95% cost savings compared to solutions based on data center GPUs.
Are you running more than $250K/yr in MDS compute? Migrate to the lowest cost GPU cloud with free, white-glove engineering support.
Why run Molecular Simulations on GPUs?
GPUs have a high degree of parallelism, which means they can perform many calculations simultaneously. This is particularly useful for molecular simulations, which involve many repetitive calculations, such as evaluating forces between atoms. Using GPUs can significantly accelerate molecular simulations, offering nearly real-time feedback and allowing researchers to run more simulations in less time. This enhanced efficiency accelerates the pace of discovery and lowers computational costs.
OpenMM benchmark methodology
The OpenMM team has provided benchmarking code in Python, along with benchmarks of simulation speed for typical biochemical systems based on OpenMM 8.0. To conduct the benchmarking test, you can run the following scripts on the target environment:
# CPU test using typical biochemical systems
python benchmark.py --platform=CPU --seconds=60 --style=table --test=pme,apoa1pme,amber20-cellulose,amber20-stmv,amoebapme
# GPU test using typical biochemical systems
python benchmark.py --platform=CUDA --seconds=60 --style=table
--test=pme,apoa1pme,amber20-cellulose,amber20-stmv,amoebapme
Following the OpenMM benchmarks, we used OpenMM 8.0 with CUDA 11.8 to build the container image. When running on SaladCloud, it first executes the benchmarking code, reports the test data to an AWS DynamoDB table, and then exits. Finally, the data is downloaded and analyzed using Pandas on JupyterLab.
We primarily focused on two key performance indicators across three scenarios:
ns/day stands for nanoseconds per day. It measures simulation speed, indicating how many nanoseconds of simulated time can be computed in one day of real-time.
ns/dollar stands for nanoseconds per dollar. It measures cost-effectiveness, showing how many nanoseconds of simulated time can be computed for one dollar.
Molecular simulations often operate on the timescale of nanoseconds to microseconds, as molecular motions and interactions occur very rapidly.
Below are the three scenarios and the methods used to collect data and calculate the final results:
| Scenario | Resource | Simulation Speed (ns/day) | Cost Effectiveness (ns/dollar) |
|---|---|---|---|
| CPUs | 16 vCPUs 8GB RAM | Create a container group with 100 instances with all GPU types on SaladCloud and run it for a few hours. Collect test data from thousands of unique Salad nodes, ensuring sufficient samples for each GPU type. Calculate the average performance for each GPU type. | Pricing from the SaladCloud Price Calculator: $0.040/hour for 8 vCPUs, 8GB RAM $0.072/hour for 16 vCPUs, 8GB RAM $0.02 ~ $0.30/hour for different GPU types https://salad.com/pricing |
| Consumer GPUs | 8 vCPUs 8GB RAM 20+ GPU types | Create a container group with 100 instances with all GPU types on SaladCloud and run it forofew hours. Collect test data from thousands of unique Salad nodes, ensuring sufficient samples for each GPU type. Calculate the average performance for each GPU type. | Pricing from the SaladCloud Price Calculator: $0.040/hour for 8 vCPUs, 8GB RAM $0.072/hour for 16 vCPUs, 8GB RAM $0.02 ~ $0.30/hour for different GPU types https://salad.com/pricing |
| Datacenter GPUs | A100 H100 | Use the test data in the OpenMM benchmarks. | Pricing from the AWS EC2 Capacity Blocks: $1.844/hour for 1 A100 $4.916/hour for 1 H100 https://aws.amazon.com/ec2/capacityblocks/pricing/ |
It is worth mentioning that performance can be influenced by many factors, such as operating systems (Windows, Linux, or WSL) and their versions, CPU models, GPU models, and driver versions, CUDA framework versions, OpenMM versions, and additional features enabled in the runtime environment. It is very common to see different results between our benchmarks and those of others.
Benchmark Results
Here are five typical biochemical systems used to benchmark OpenMM 8.0, along with the corresponding test scripts:
| Model | Description | Test script | |
|---|---|---|---|
| 1 | Dihydrofolate Reductase (DHFR), Explicit-PME | This is a 159 residue protein with 2489 atoms. The version used for explicit solvent simulations included 7023 TIP3P water molecules, giving a total of 23,558 atoms. All simulations used the AMBER99SB force field and a Langevin integrator. | python benchmark.py –platform=CUDA or CPU –seconds=60 –test=pme |
| 2 | Apolipoprotein A1 (ApoA1), PME | This consists of 392 protein residues, 160 POPC lipids, and 21,458 water molecules, for a total of 92,224 atoms. All simulations used the AMBER14 force field. | python benchmark.py –platform=CUDA or CPU –seconds=60 –test=apoa1pme |
| 3 | Cellulose, PME | It consists of a set of cellulose molecules (91,044 atoms) solvated with 105,855 water molecules, for a total of 408,609 atoms. | python benchmark.py –platform=CUDA or CPU –seconds=60 –test=amber20-cellulose |
| 4 | Satellite Tobacco Mosaic Virus (STMV), PME | It consists of 8820 protein residues, 949 RNA bases, 300,053 water molecules, and 649 sodium ions, for a total of 1,067,095 atoms. | python benchmark.py –platform=CUDA or CPU –seconds=60 –test=amber20-stmv |
| 5 | AMOEBA DHFR, PME | Full mutual polarization was used, with induced dipoles iterated until they converged to a tolerance of 1e-5. | python benchmark.py –platform=CUDA or CPU –seconds=60 –test=amoebapme |
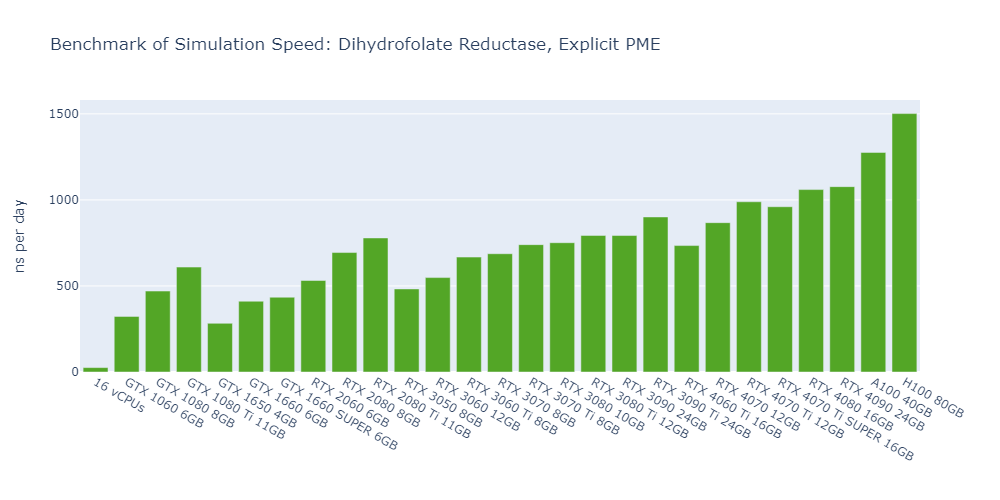
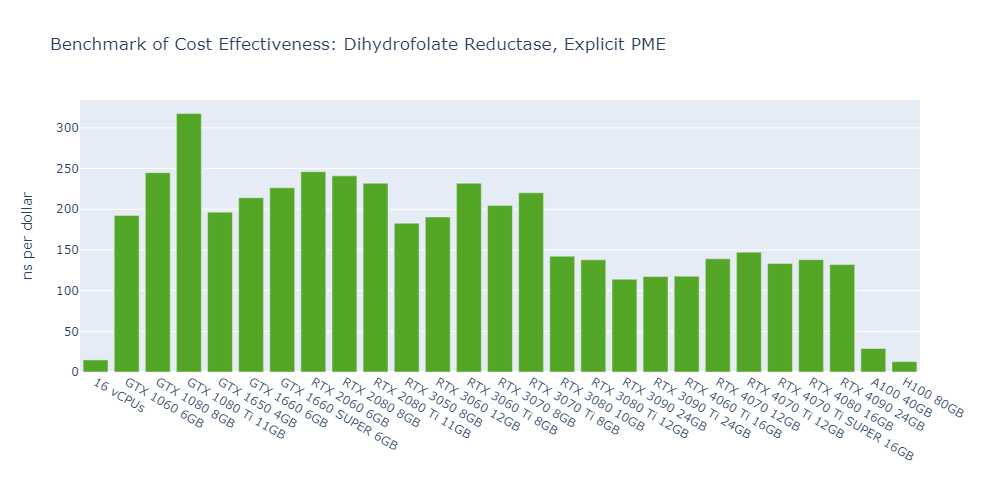
Model 1: Dihydrofolate Reductase (DHFR), Explicit-PME
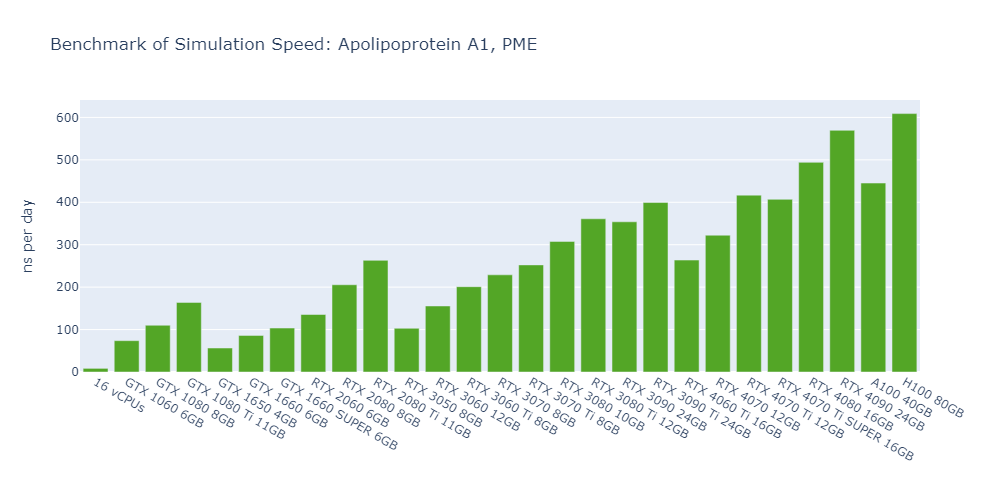
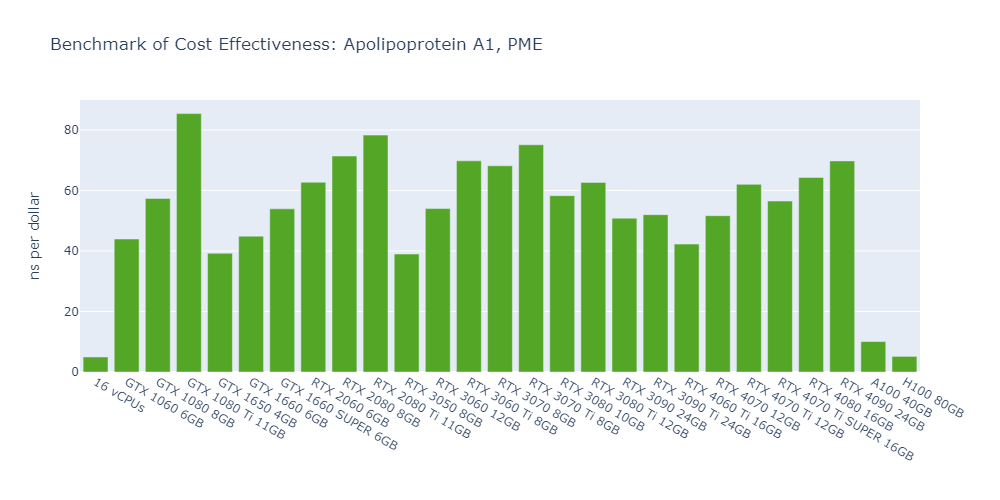
Model 2: Apolipoprotein A1 (ApoA1), PME
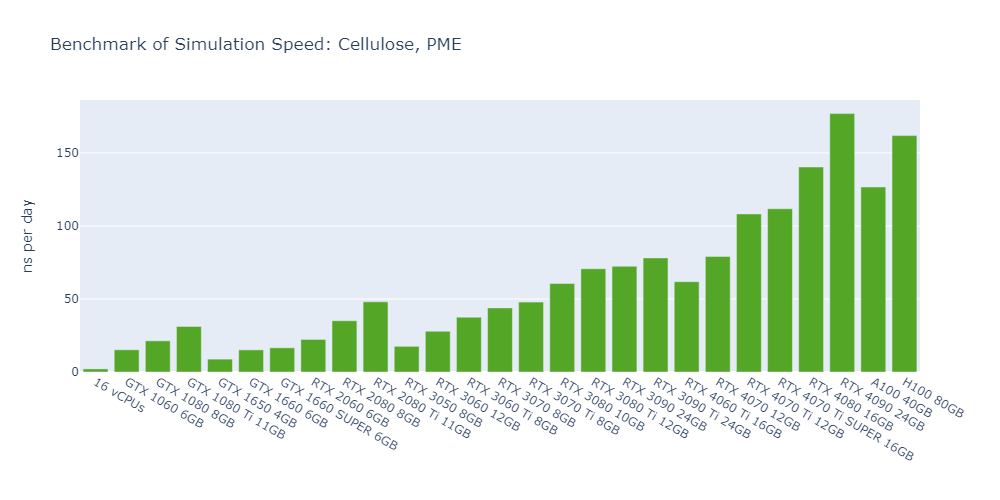
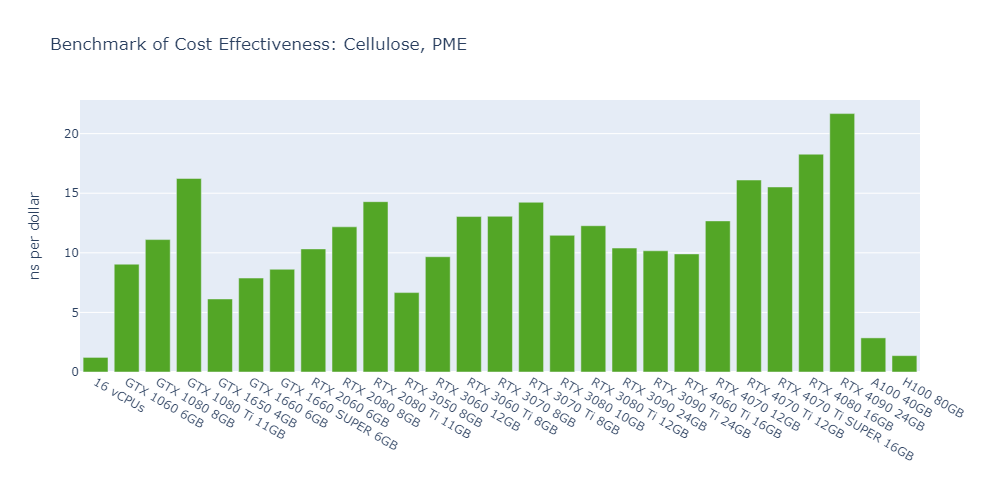
Model 3: Cellulose, PME
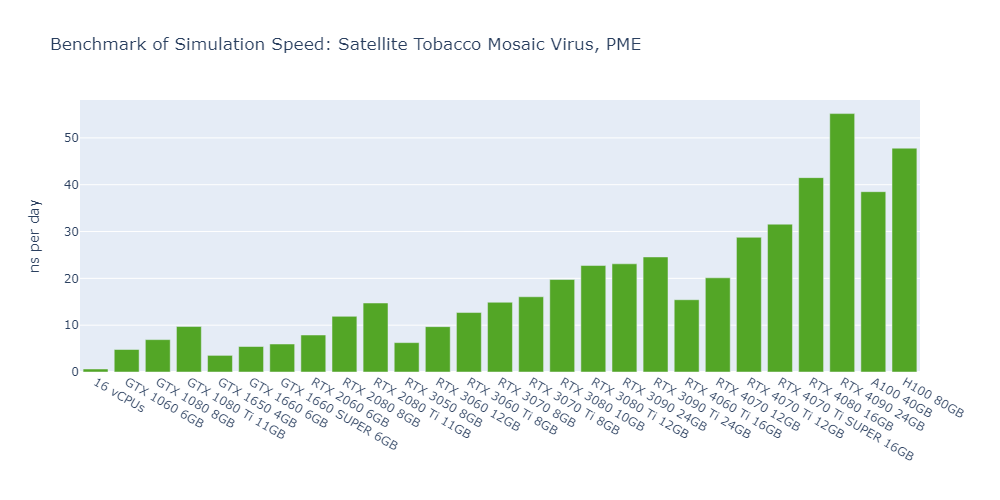
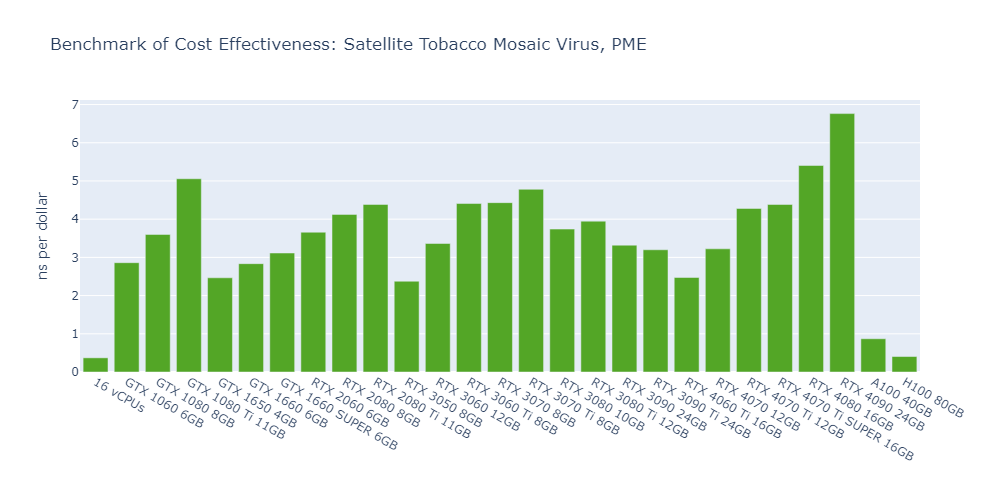
Model 4: Satellite Tobacco Mosaic Virus (STMV), PME
Model 5: AMOEBA DHFR, PME
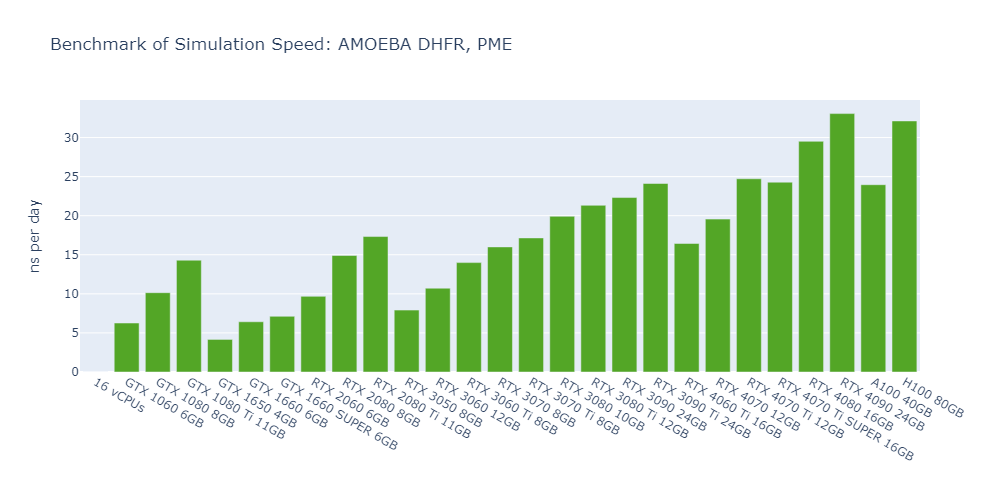
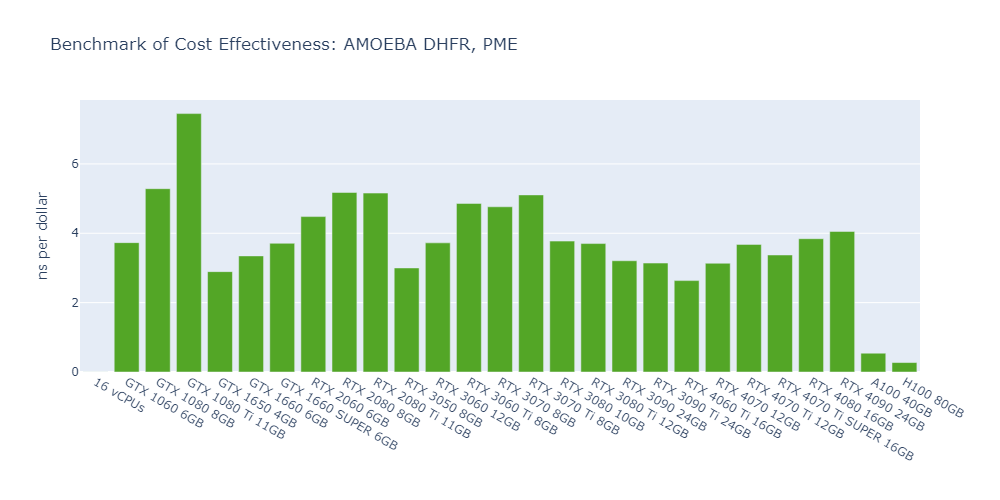
Observations from the OpenMM GPU benchmarks:
Here are some interesting observations from the OpenMM GPU benchmarks:
The VRAM usage for all simulations is only 1-2 GB, which means nearly all platforms (CPU-only or GPU) and all GPU types can theoretically be utilized to run these models.
For all models, the simulation speed of GPUs is significantly higher than that of CPUs, ranging from nearly hundreds of times in Model 1 to more than tens of thousands of times in Model 5.
In general, high-end GPUs outperform low-end GPUs in terms of simulation speed. However, the flagship model of a given GPU family often surpasses the low-end models of the next family.
As models become more complex with additional molecules and atoms, the performance differences between low-end GPUs and high-end GPUs become more pronounced, ranging from a few times to tens of times. For example, in Model 1, the H100 outperforms the 1650 by 530%, while in Model 3, the 4090 surpasses the 1650 by 2000%.
In 3 out of 5 models, the consumer GPUs (4080, 4090) outperform the data center GPUs (A100, H100).
CPU-only platforms are not suitable for molecular simulations, not only because of their extremely low performance but also due to their high cost relative to that performance.
The cost-effectiveness of data center GPUs (A100, H100) is also very low for molecular simulation jobs, close to that of CPU-only platforms. In Models 1 and 5, the 1080 Ti surpasses the H100 by 2500% in terms of cost-effectiveness.
The fact that molecular simulation jobs require little VRAM doesn’t mean low-end GPUs are always the better choice for cost savings than high-end GPUs. For example, the most cost-effective GPU in Models 1, 2, and 5 is the 1080 Ti, while the 4090 ranks as the top performer in Models 3 and 4.
Given the complexity and variety of these biochemical systems, the best-performing GPU and the most cost-effective GPU for each model might differ. Before deploying your workloads extensively on SaladCloud, we recommend conducting a quick benchmarking test to identify the optimal resource types for your models. This will allow you to optimize your workloads for reduced costs, improved performance, or both.
In need of a more cost-effective GPU cloud for simulation scalability? Migrate to SaladCloud, covering up to 100TB of egress fees for limited time.
Reference Design for Molecular Simulation Workloads on SaladCloud
Molecular simulations typically involve running thousands of jobs across various models and parameters. Each job can take several hours or more, depending on the complexity and the selected resource types. To enhance the system throughput, reliability, and efficiency, the proposed batch-processing architecture for molecular simulations comprises the following components:
- Cloud Storage: The initial input, intermediate checkpoint and final output of each job are organized and stored using either three separate folders or a single folder with different files in Cloudflare R2, which is AWS S3 compatible and incurs zero egress fees.
- Job Queue System: The job queue, such as Salad Job Queue, Salad Kelpie, AWS SQS or others, is to manage and keep the jobs for the Salad nodes to retrieve and process.
- Job Creator: After creating the folder(s) in Cloud Storage and uploading the initial input (such as an OpenMM PDB file, that defines the molecular structure of a biochemical system), it wraps this information into a job and submits the job to the Job Queue. The Job Creator can provide a webhook for callback when the job is done, or checks the job status or result directly. Once the job is completed, the Job Creator can download the job output for further analysis and utilization.
- GPU Resource Pool: Hundreds of Salad nodes equipped with dedicated GPUs are utilized for tasks such as pulling a job, downloading its input and checkpoint, running the simulation, uploading the checkpoint regularly, and uploading the final result when the job is done.
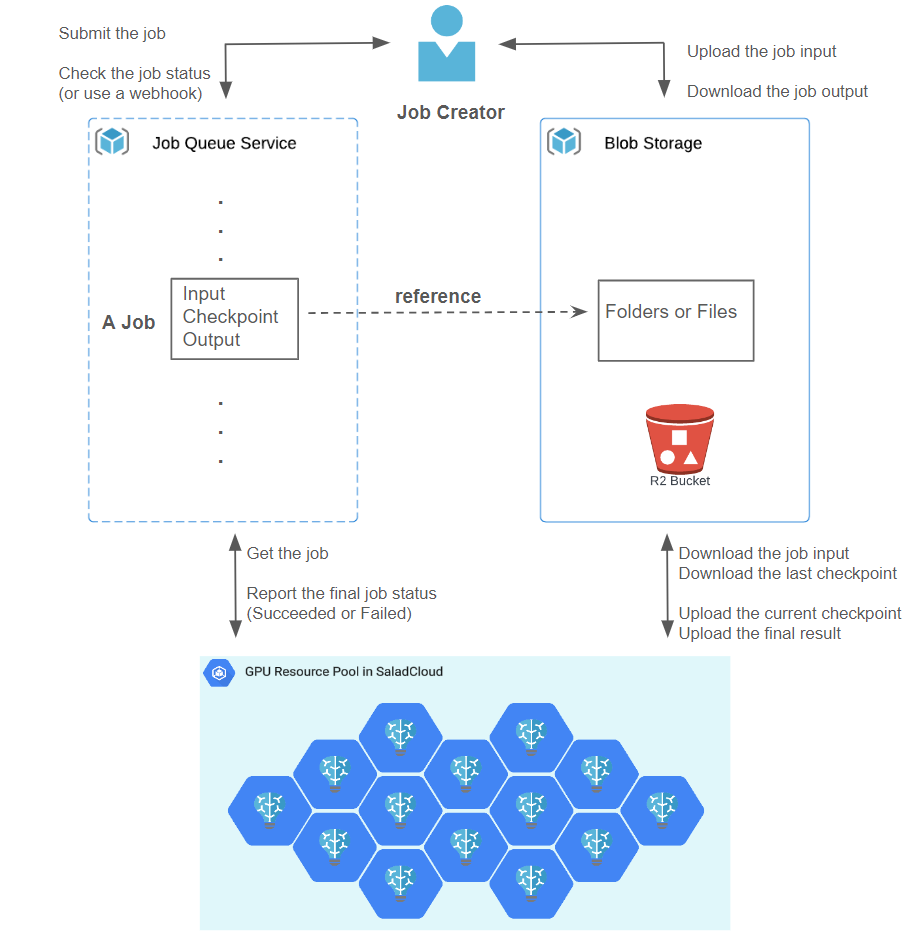
SaladCloud operates on a foundation of shared nodes, where a node might go down while a job runs. A new node will be allocated in such cases, and the unfinished job will be re-queued in the Job Queue. To prevent a job from being processed from scratch after node reallocation, the code needs to include some logic:
Step 1: When retrieving a job, first download both the job input and checkpoint from the cloud; if there is no checkpoint, run the simulation from scratch; otherwise, continue the job execution from the last checkpoint.
Step 2: After completing some simulation steps or running for a certain period (such as 10 minutes), make the latest checkpoint and upload it to the cloud.
Step 3: When the job is finished, upload the final result to the cloud and return the status to the Job Queue.
Molecular simulations are typically compute-bound tasks, and the input, checkpoint, and output files sizes are small (a few MB), resulting in minimal upload and download times. Implementing a dedicated I/O thread to regularly upload the checkpoint can help eliminate any impact on simulation throughput.
You can implement the proposed solution using Salad Job Queue or AWS SQS.
If you don’t want to deal with data synchronization in your code (such as downloading input and checkpoint, uploading checkpoint and output), Salad Kelpie can be a good choice. It provides a job queue service where you can submit a job using its APIs. The Kelpie worker, integrated with your workload running on Salad nodes, can retrieve a job, execute your code, and handle all data synchronization in the background for you between local folders/files and S3-compatible Cloud Storage.
Here is an example of an OpenMM-based simulation application integrated with the Kelpie worker:
https://github.com/SaladTechnologies/openmm
SaladCloud: The right choice for Molecular Dynamics Simulations
The OpenMM GPU benchmark shows that consumer GPUs are the right choice for running molecular simulation jobs. They offer performance comparable to data center GPUs while delivering over 95% cost savings. With the right design and implementation, we can build a high-throughput, reliable, and cost-effective system on SaladCloud to run simulation jobs at a very large scale.
If your needs involve running millions of simulation jobs quickly or requiring access to hundreds of GPUs or various GPU types within minutes for testing or research purposes, SaladCloud is the ideal platform.
Are you running more than $250K/yr in MDS compute? Migrate to the lowest cost GPU cloud with 1:1 prepaid credit matching from SaladCloud!
SaladCloud is the world’s largest distributed cloud computing network with 11,000+ daily GPUs and 450,000 GPUs contributing compute, all at the lowest cost in the market.


